
ABSTRAK
Latar belakang
Mutasi POU4F3 menyebabkan DFNA15, gangguan pendengaran nonsindromik dominan autosomal. POU4F3 mengkodekan faktor transkripsi yang penting untuk perkembangan dan pemeliharaan sel rambut telinga bagian dalam.
Tujuan
Untuk mengidentifikasi dan mengkarakterisasi secara fungsional mutasi POU4F3 baru pada dua keluarga Tiongkok dengan gangguan pendengaran progresif yang terjadi pada usia lanjut.
Metode
Pengurutan DNA paralel secara masif (MPS) dilakukan pada individu yang terkena dampak dari dua keluarga Tionghoa yang tidak berhubungan. Pengurutan Sanger memvalidasi mutasi dan mengonfirmasi ko-segregasi. Analisis fungsional meliputi analisis ekspresi protein dengan Western blot dan studi lokalisasi subseluler dengan imunofluoresensi.
Hasil
Kami mengidentifikasi dua mutasi nonsense baru pada POU4F3 : c.863C > A (p.Ser288Ter) dan c.172G > T (p.Glu58Ter), keduanya berkosegregasi dengan fenotipe gangguan pendengaran. Studi fungsional menunjukkan p.Ser288Ter menghasilkan protein yang stabil tetapi salah lokasi dengan gangguan transpor nukleus, sementara p.Glu58Ter menghasilkan protein yang sangat terpotong dan cepat terdegradasi.
Kesimpulan
Studi ini memperluas spektrum mutasi DFNA15 dan memberikan wawasan baru mengenai patogenesis gangguan pendengaran terkait POU4F3. Temuan kami menunjukkan bahwa mekanisme molekuler yang berbeda dapat menghasilkan fenotipe DFNA15 yang serupa, mendukung haploinsufisiensi POU4F3 sebagai mekanisme patogenik utama.
1 Pendahuluan
Gangguan pendengaran adalah salah satu cacat sensorik yang paling umum, yang memengaruhi jutaan orang di seluruh dunia (Chadha et al. 2021 ). Faktor genetik memainkan peran penting dalam etiologi gangguan pendengaran. Lebih dari 50% kasus gangguan pendengaran bawaan disebabkan oleh penyebab genetik (Morton dan Nance 2006 ). Gangguan pendengaran genetik dapat diklasifikasikan sebagai sindromik, di mana gangguan pendengaran dikaitkan dengan fitur klinis lainnya, atau nonsindromik, di mana gangguan pendengaran adalah satu-satunya manifestasi klinis. Gangguan pendengaran nonsindromik (NSHL) mencakup sekitar 70% kasus gangguan pendengaran genetik dan dapat diwariskan dalam pola autosom dominan, autosom resesif, terkait-X, atau mitokondria (Walls et al. 2024 ). Hingga saat ini, lebih dari 150 gen telah diidentifikasi sebagai penyebab NSHL, yang menyoroti heterogenitas genetik dari kondisi ini (Shearer et al. 2023 ).
Di antara gen yang terlibat dalam NSHL, POU4F3 (602.460, domain POU, kelas 4, faktor transkripsi 3) telah muncul sebagai pemain penting dalam perkembangan dan fungsi telinga bagian dalam (Vahava et al. 1998 ). POU4F3 , juga dikenal sebagai BRN3C , adalah anggota keluarga faktor transkripsi POU dan diekspresikan dalam sel-sel rambut koklea dan sistem vestibular (Xiang et al. 1997 ). POU4F3 memainkan peran penting dalam diferensiasi dan kelangsungan hidup sel-sel rambut telinga bagian dalam, yang penting untuk transduksi dan keseimbangan suara (Erkman et al. 1996 ). Mutasi dalam POU4F3 telah dikaitkan dengan NSHL DFNA15 autosomal dominan, yang ditandai dengan gangguan pendengaran progresif yang biasanya dimulai pada dekade kedua hingga ketiga kehidupan (Collin et al. 2008 ). Mekanisme yang mendasari patogenesis DFNA15 rumit dan tidak sepenuhnya dijelaskan. Namun, penelitian telah menunjukkan bahwa mutasi POU4F3 dapat menyebabkan gangguan aktivitas transkripsi, perubahan lokalisasi protein, atau penurunan stabilitas protein (Weiss et al. 2003 ), yang mengakibatkan haploinsufisiensi protein POU4F3 (Zhu et al. 2020 ). Cacat molekuler ini dapat mengakibatkan stereosilia sel rambut progresif dan disfungsi mitokondria serta degenerasi sel rambut, yang pada akhirnya menyebabkan gangguan pendengaran (Hertzano et al. 2004 ; Zhu et al. 2020 ). Variabilitas fenotipik yang diamati pada pasien DFNA15, termasuk perbedaan usia onset, laju perkembangan, dan tingkat keparahan gangguan pendengaran, menunjukkan bahwa faktor genetik atau lingkungan tambahan dapat memengaruhi perjalanan penyakit (Lee et al. 2010 ; Zhu et al. 2020 ).
Meskipun pemahaman tentang peran POU4F3 dalam gangguan pendengaran semakin berkembang, spektrum penuh mutasi patogenik pada gen ini masih harus dijelaskan. Dalam penelitian ini, kami melaporkan dua mutasi nonsense baru pada gen POU4F3 yang diidentifikasi dalam dua keluarga Tiongkok dengan NSHL progresif yang terjadi pada akhir hayat dan memberikan karakterisasi fungsional dari mutasi ini pada ekspresi dan lokalisasi protein POU4F3. Temuan ini tidak hanya memperluas spektrum mutasi DFNA15 tetapi juga memberikan wawasan baru tentang mekanisme patogenik gangguan pendengaran terkait POU4F3. Hasil kami berkontribusi pada pengetahuan yang berkembang tentang DFNA15 dan mungkin memiliki implikasi untuk strategi diagnostik dan terapi masa depan untuk gangguan pendengaran turunan.
2 Bahan dan Metode
2.1 Subjek dan Evaluasi Klinis
Dua keluarga, NT-107 dan NT-213, dengan gangguan pendengaran dominan autosomal telah tinggal di Provinsi Jiangsu, Tiongkok selama lebih dari empat generasi. Peserta menjalani evaluasi pendengaran komprehensif, termasuk audiometri nada murni (PTA), pemeriksaan otoskopi, dan pemindaian CT resolusi tinggi tulang temporal bila perlu. Riwayat medis terperinci dikumpulkan, termasuk usia onset, tingkat perkembangan, penggunaan alat bantu dengar, paparan kebisingan, gejala vestibular, dan manifestasi sindromik lainnya. Tidak ada individu yang direkrut yang memiliki diagnosis sindromik. PTA dihitung dari ambang batas audiometrik pada empat frekuensi (0,5, 1, 2, dan 4 kHz). Tingkat gangguan pendengaran dikategorikan ke dalam lima kelompok berdasarkan rata-rata konduksi udara nada murni: ringan (26–40 dB HL), sedang (41–55 dB HL), cukup parah (56–70 dB HL), parah (71–90 dB HL), dan sangat parah (> 91 dB HL). Penelitian ini disetujui oleh Komite Etik Rumah Sakit Afiliasi Universitas Nantong (2023-L115). Persetujuan tertulis diperoleh dari semua peserta atau orang tua mereka.
2.2 Pengurutan DNA Paralel Massal
DNA genom diekstraksi dari sampel darah tepi proband dan anggota keluarga mereka (bila tersedia) menggunakan kit DNA Darah (Tiangen Biotech, Tiongkok). Proband (IV-2) dari keluarga NT-107 dan proband (III-5) dari keluarga NT-213 menjalani pengurutan DNA paralel masif (MPS) yang ditargetkan yang mencakup 270 gen yang terkait dengan ketulian (Tabel S1 ). Varian dipilih melalui analisis genetik bertahap seperti yang dijelaskan sebelumnya (Wu et al. 2020 ; Zhang et al. 2024 ). Data pengurutan dianalisis berdasarkan pola pewarisan dominan autosom (AD). Varian kandidat dikonfirmasi melalui pengurutan Sanger pada semua anggota keluarga yang tersedia, dan patogenisitasnya ditafsirkan menggunakan standar dan pedoman American College of Medical Genetics (ACMG). Pengurutan Sanger dilakukan dengan menggunakan primer: Maju 5′-TCTGATCCACACGTCTGTTCC-3′ dan Mundur 5′-AGGTGGTGTGGATGGATCTG-3′ untuk mutasi c.172G > T; Maju 5′-GAAGAACAGCAAGCCAGAGC-3′ dan Mundur 5′-GATGAAGGACGTGGCTGGAT-3′ untuk mutasi c.863C > A.
2.3 Konstruksi Plasmid
Tikus tipe liar Pou4f3 (NM_138945.2), p.Glu58Ter, dan varian p.Ser288Ter dengan Flag-tagged di ujung N-nya dikloning ke dalam plasmid pcDNA3.1 atau pIRES-EGFP, menggunakan situs EcoRV (R3195V, New England Biolabs), EcoRI (R3101S, New England Biolabs), dan BamhI (R3136S, New England Biolabs). Bakteri DH5α digunakan untuk mengamplifikasi semua plasmid ekspresi tikus. Urutan plasmid divalidasi dengan sekuensing Sanger.
2.4 Kultur Sel dan Transfeksi
Larutan penisilin-streptomisin (E607011, Sangon Biotech), asam amino nonesensial (NEAA, 11140050, Gibco), dan 10% serum sapi janin (FBS, 40130ES76, Yeasen) ditambahkan ke DMEM (12.800.017, Gibco) untuk mendukung pertumbuhan sel HEK293T (ATCC: CRL-3216). Reagen Transfeksi Liposomal Hieff TransTM (40802ES03, Yeasen) digunakan untuk mentransfeksi sel HEK293T dalam Opti-MEM (31.985.062, Thermo Fisher). Sel dipanen 24 jam setelah transfeksi untuk analisis lebih lanjut.
2.5 Ekstraksi RNA dan RT-qPCR
Setelah dua putaran pencucian dengan 1 mL larutan penyangga fosfat (PBS), 0,5 mL reagen RNAiso Plus (9109, Takara) ditambahkan ke sel HEK293T. Sesuai petunjuk produsen, total RNA sampel diekstraksi. HiScript III RT SuperMix (+gDNA wiper) (R323-01, Vazyme) digunakan untuk melakukan transkripsi balik (RT) pada total sampel RNA. AceQ qPCR SYBR Green Master Mix (Q111-02, Vazyme) digunakan untuk PCR kuantitatif (qPCR) menggunakan instrumen Roche Cycler 96. Primer yang digunakan dalam percobaan qPCR adalah: untuk Pou4f3, 5′-TCTGGCGGCGGTGGATAT-3′ dan 5′-GCTGCTCATGGTATGGTAGGT-3′ dan untuk Gapdh, 5′-ACCACGAGAAATATGACAACTCAC-3′ dan 5′-CCAAAGTTGTCATGGATGACC-3′. Gen referensi internal Gapdh digunakan untuk menormalkan ekspresi gen target.
2.6 Analisis Western Blot
Sel yang dikultur dibilas dua kali dengan PBS. Sel diekstraksi dan dihomogenkan dengan buffer lisis, 150 mM NaCl, 50 mM Tris–HCl pH 7,4, 1% Triton X-100 (A110694-0500, Sangon Biotech), 1 mM etilen diamina tetra-asam asetat pH 8,0, dan 1 mM fenilmetilsulfonil fluorida (ST005, Beyotime) yang mengandung Protease Inhibitor Cocktail lengkap (11,697,498,001, Roche) pada suhu 4°C selama 10 menit. Protein dikenakan SDS–PAGE dan dipindahkan ke membran PVDF dan dianalisis dengan antibodi berikut: antibodi anti-Flag tikus (1:2000, 30503ES60, Yeasen), anti-Actin tikus (1:20000, 4970, Cell Signaling Technology), dan antibodi anti-GAPDH tikus (BioWorld, MN, AS), diikuti oleh inkubasi dalam antibodi sekunder HRP anti-tikus (1:5000, BS12478, Bioworld) selama 2 jam pada suhu ruangan (RT). Sinyal divisualisasikan menggunakan substrat ECL (180–5001, Tanon) pada sistem analisis citra chemiluminescence/fluorescence otomatis (Tanon 4600, Tanon).
2.7 Imunofluoresensi Wholemount
Setelah difiksasi di atas es dalam 4% PFA selama 30 menit, dibilas dua kali dengan PBS, sel yang ditransfeksi diblokir dalam 5% serum kuda normal (NHS) dengan 0,3% Triton X-100 dalam PBS selama 1 jam pada suhu kamar. Sampel kemudian diinkubasi semalam pada suhu 4°C dengan antibodi primer (Anti-Flag, Cell Signaling Technology, 14.793) yang telah diencerkan dalam 1% NHS dengan 0,3% Triton X-100 dalam PBS. Sampel diinkubasi dengan antibodi sekunder goat anti-rabbit IgG (H + L) (Jackson ImmunoResearch, Inggris) selama 2 jam pada suhu RT. Sampel dicitrakan oleh Leica SP5-II (Leica, Jerman). Perangkat lunak ImageJ (versi 1.46r, NIH, MD) digunakan untuk pemrosesan citra z-stack confocal.
3 Hasil
3.1 Fenotipe Audiologis dan Penilaian Klinis
Silsilah keluarga NT-107 (lima generasi) dan NT-213 (empat generasi) menunjukkan pewarisan dominan autosomal (Gambar 1A,B ). Semua individu yang terpengaruh menunjukkan kehilangan pendengaran sensorineural nonsindromik (NSHL), yang bilateral dan simetris, mulai dari sedang hingga sangat dalam. Kehilangan pendengaran biasanya dimulai pada frekuensi tinggi dan berkembang menjadi konfigurasi audiometrik yang menurun. Usia onset berbeda antara keluarga: dekade ketiga untuk NT-107 dan akhir dekade kedua untuk NT-213. Kemunduran pendengaran terjadi di semua frekuensi, berkembang menjadi kehilangan pendengaran tipe datar yang parah setelah usia 60 tahun pada NT-107 dan usia 50 tahun pada NT-213. Keluarga NT-213 menunjukkan kehilangan pendengaran yang lebih parah dibandingkan dengan NT-107, berkorelasi dengan durasi kehilangan pendengaran. Karakteristik fenotipe pendengaran diilustrasikan dalam Gambar 1C,D .
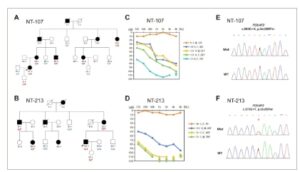
GAMBAR 1
Buka di penampil gambar
Kekuatan Gambar
Analisis fenotipik dan genetik untuk kedua keluarga. (A, B) Silsilah dari dua keluarga independen ditunjukkan. (C, D) Audiogram representatif dari anggota dengan gangguan pendengaran dari Keluarga NT-107 dan NT-213. (E, F) Hasil sekuensing dari keluarga NT-107 dan NT-213. Perhatikan bahwa NT-107:V-1 dan NT-213:IV-1 masing-masing berusia 12 dan 9 tahun, dan keduanya ditandai sebagai tidak terpengaruh.
3.2 Identifikasi Kandidat Mutasi
Analisis ko-segregasi dari mutasi kandidat yang diidentifikasi oleh MPS dilakukan pada anggota keluarga yang tersedia. Dalam keluarga NT-107, analisis terperinci dari alel umum di antara lima anggota keluarga yang terpengaruh mengungkapkan varian heterozigot baru POU4F3 (NM_002700.3): c.863C > A, p.Ser288Ter. Substitusi C ke A pada posisi nukleotida 863 ini memperkenalkan kodon stop prematur pada asam amino 288. Dalam keluarga NT-213, mutasi POU4F3 baru: c.172G > T, p.Glu58Ter (NM_002700.3) diidentifikasi pada empat individu yang terpengaruh tetapi tidak ada pada anggota keluarga normal yang berusia lebih dari 30 tahun. Transisi G ke T ini pada posisi nukleotida 172 juga menciptakan kodon stop prematur pada asam amino 58. Kedua mutasi heterozigot pada individu yang terpengaruh ini divalidasi oleh sekuensing Sanger (Gambar 1E,F ). Kedua mutasi nonsense tidak ditemukan dalam basis data gnomAD dan ClinVar. Mengikuti rekomendasi ACMG, kedua mutasi POU4F3 , p.Glu58Ter dan p.Ser288Ter, diklasifikasikan sebagai kemungkinan patogen. Kami juga mengidentifikasi c.863C > A pada satu keturunan yang tidak terpengaruh dalam famili NT-107 (V-1) dan c.172G > T dalam famili NT-213 (IV-1). Meskipun kedua subjek (masing-masing berusia 13 dan 11 tahun) dilaporkan sehat, mereka mungkin mengalami kehilangan pendengaran progresif di usia selanjutnya.
3.3 Analisis Fungsional Dua Mutasi Baru POU4F3
Untuk mengeksplorasi konsekuensi fungsional dari dua mutasi POU4F3 baru, kami membangun plasmid yang mengekspresikan protein POU4F3 tipe liar (WT) yang ditandai Flag, p.Ser288Ter (S288X) dan p.Glu58Ter (E58X).
Pertama, mutasi S288X yang diidentifikasi dari famili NT-107 menghasilkan kodon stop prematur yang menghasilkan protein POU4F3 terpotong dalam domain homeo POU (Gambar 2A ). Analisis western blot pada sel HEK293 yang ditransfeksi menunjukkan bahwa sementara mutan S288X POU4F3 mudah diekspresikan, ukurannya lebih kecil daripada kontrol tipe liar (Gambar 2B ). Baik transkrip mRNA POU4F3 WT maupun S288X diekspresikan pada tingkat yang setara (Gambar 2C ). Menariknya, sementara protein POU4F3 WT terlokalisasi ke nukleus seperti yang diharapkan, protein POU4F3 mutan S288X terdistribusi di nukleus dan sitosol (Gambar 2D ). Hasil-hasil ini menunjukkan bahwa mutan S288X mungkin telah merusak lokalisasi nuklir POU4F3, konsisten dengan sinyal lokalisasi nuklir yang diduga berada di ujung C POU4F3 (Weiss et al. 2003 ).
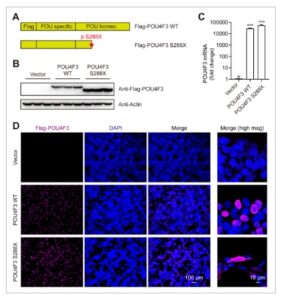
GAMBAR 2
Buka di penampil gambar
Kekuatan Gambar
Protein mutan POU4F3 S288X salah terlokalisasi ke sitoplasma dalam sel HEK293T. (A) Ilustrasi skematis dari struktur protein POU4F3 tipe lebar (WT) dan mutan (S288X) berlabel Flag. Bintang menunjukkan lokasi mutasi. (B) Ekspresi protein Flag-POU4F3 WT atau Flag-POU4F3 S288X dalam sel HEK293T yang ditransfeksi melalui western blotting. (C) mRNA dari konstruksi Flag-POU4F3 WT dan Flag-POU4F3 S288X dideteksi oleh RT-qPCR dalam sel HEK293T yang ditransfeksi. *** p < 0,001 melalui uji- t Student yang tidak berpasangan . (D) Gambar imunofluoresensi Flag-POU4F3 dalam sel HEK293T yang ditransfeksi dengan vektor kosong, Flag-POU4F3 WT, atau plasmid Flag-POU4F3 S288X.
Kedua, mutasi E58X yang diidentifikasi dari famili NT-213 mengakibatkan terminasi prematur dalam domain spesifik POU terminal-N (Gambar 3A ). Seperti yang diharapkan, protein mutan E58X gagal diekspresikan secara stabil dalam sel HEK293 yang ditransfeksi (Gambar 3B ), meskipun mRNA mutan tersebut diekspresikan (Gambar 3C ). Secara konsisten, hasil imunofluoresensi juga menunjukkan kurangnya ekspresi protein mutan E58X POU4F3 yang stabil, sementara protein EGFP dari plasmid pIRES mudah dideteksi (Gambar 3D ). Hasil-hasil ini menunjukkan bahwa mutasi E58X mengakibatkan degradasi signifikan protein POU4F3 yang terpotong.
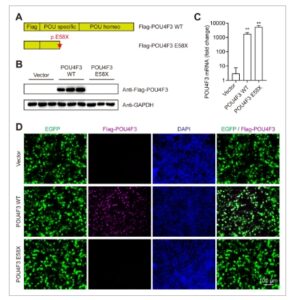
GAMBAR 3
Buka di penampil gambar
Kekuatan Gambar
Protein mutan POU4F3 E58X gagal diekspresikan dalam sel HEK293T. (A) Ilustrasi skematis dari struktur protein POU4F3 tipe lebar (WT) dan mutan (E58X) berlabel FLAG. Bintang menunjukkan lokasi mutasi. (B) Ekspresi protein Flag-POU4F3 WT atau Flag-POU4F3 E58X dalam sel HEK293T yang ditransfeksi melalui western blotting. (C) mRNA dari konstruksi Flag-POU4F3 WT dan Flag-POU4F3 E58X dideteksi oleh RT-qPCR dalam sel HEK293T yang ditransfeksi. ** p < 0,01 melalui uji- t Student yang tidak berpasangan . (D) Gambar imunofluoresensi Flag-POU4F3 dalam sel HEK293T yang ditransfeksi dengan vektor kosong, Flag-POU4F3 WT, atau plasmid Flag-POU4F3 E58X. Protein EGFP diekspresikan bersama dari vektor pIRES-ERFP. Bersama-sama, mutasi E58X dan S288X mengakibatkan degradasi dan mislokalisasi protein POU4F3, yang kemungkinan mengganggu aktivitas transkripsi POU4F3. 4 Diskusi Dalam penelitian ini, kami mengidentifikasi dan mengkarakterisasi secara fungsional dua mutasi nonsense baru pada gen POU4F3 (c.863C > A, p.Ser288Ter dan c.172G > T, p.Glu58Ter) pada dua keluarga Tionghoa dengan NSHL progresif yang terjadi pada akhir masa pertumbuhan. Temuan ini tidak hanya memperluas spektrum mutasi DFNA15 tetapi juga memberikan wawasan baru mengenai mekanisme patogenik dari gangguan pendengaran terkait POU4F3.
Perluasan spektrum mutasi POU4F3 sangat penting karena beberapa alasan. Pertama, hal ini meningkatkan pemahaman kita tentang korelasi genotipe-fenotipe dalam DFNA15, yang berpotensi menjelaskan variabilitas dalam usia onset dan tingkat perkembangan yang diamati di antara pasien (Lee et al. 2023 ). Kedua, mengidentifikasi mutasi baru berkontribusi untuk meningkatkan diagnosis genetik dan konseling bagi keluarga yang terkena gangguan pendengaran herediter (Usami et al. 2002 ). Ketiga, mengkarakterisasi mutasi baru dan konsekuensi fungsionalnya dapat memberikan wawasan tentang mekanisme molekuler yang mendasari disfungsi dan kehilangan sel rambut, yang berpotensi mengarah pada target terapi baru (Bowl dan Brown 2018 ).
Protein POU4F3 adalah faktor transkripsi penting untuk pengembangan dan pemeliharaan sel rambut telinga bagian dalam (Xiang et al. 1997 ). Analisis fungsional kami terhadap dua mutasi baru ini mengungkapkan mekanisme molekuler berbeda yang menyebabkan disfungsi POU4F3. Mutasi p.Ser288Ter menghasilkan protein terpotong yang, meskipun stabil, menunjukkan lokalisasi nuklir yang terganggu. Pengamatan ini konsisten dengan penelitian sebelumnya yang menunjukkan adanya sinyal lokalisasi nuklir di C-terminus POU4F3 (Weiss et al. 2003 ). Mislokalisasi protein mutan kemungkinan membahayakan fungsinya sebagai faktor transkripsi. Sebaliknya, mutasi p.Glu58Ter menyebabkan protein terpotong parah yang cepat terdegradasi, seperti yang ditunjukkan oleh kurangnya ekspresi protein yang terdeteksi dalam analisis Western blot dan imunofluoresensi kami. Temuan ini menunjukkan bahwa mutasi ini kemungkinan besar mengakibatkan hilangnya fungsi POU4F3 secara total, mungkin melalui peluruhan mRNA mutan yang dimediasi oleh nonsense atau degradasi cepat protein yang terpotong (Mort et al. 2008 ). Konsisten dengan gagasan ini, mutasi POU4F3 lainnya telah terbukti membahayakan stabilitasnya setelah translasi (Weiss et al. 2003 ).
Menariknya, meskipun konsekuensi molekuler dari kedua mutasi ini berbeda, kedua keluarga menunjukkan fenotipe serupa dari kehilangan pendengaran progresif yang terjadi pada akhir hayat, meskipun dengan beberapa variasi dalam tingkat keparahan dan usia timbulnya. Kesamaan fenotipe ini, meskipun mekanisme molekuler yang mendasarinya berbeda, mendukung hipotesis bahwa haploinsufisiensi POU4F3 adalah penyebab utama DFNA15 (Collin et al. 2008 ; Zhu et al. 2020 ). Variabilitas dalam usia timbulnya dan tingkat perkembangan yang diamati antara kedua keluarga kami dan dibandingkan dengan kasus DFNA15 yang dilaporkan sebelumnya menunjukkan bahwa faktor genetik atau lingkungan tambahan dapat memodulasi perjalanan penyakit, seperti yang sebelumnya ditunjukkan pada model tikus DFNA15 (Zhu et al. 2020 ).
Gangguan ekspresi dan lokalisasi POU4F3 akibat mutasi ini kemungkinan akan memengaruhi ekspresi sejumlah gen yang penting bagi perkembangan dan pemeliharaan sel rambut. Penelitian sebelumnya telah mengidentifikasi sejumlah gen target POU4F3, termasuk Gfi1 dan Lhx3 , yang penting bagi diferensiasi dan kelangsungan hidup sel rambut (Hertzano et al. 2007 , 2004 ). Mutasi POU4F3 dapat menyebabkan disregulasi gen target ini, yang pada akhirnya mengakibatkan degenerasi sel rambut progresif dan kehilangan pendengaran.
Sebagai kesimpulan, penelitian kami mengidentifikasi dua mutasi nonsense baru pada POU4F3 dan menjelaskan konsekuensi molekulernya yang berbeda, sehingga memberikan wawasan baru tentang mekanisme patogenik DFNA15. Temuan ini berkontribusi pada semakin banyaknya pengetahuan tentang gangguan pendengaran DFNA15 dan dapat menginformasikan upaya masa depan dalam mengembangkan terapi yang ditargetkan untuk gangguan pendengaran genetik. Penelitian lebih lanjut yang menyelidiki efek jangka panjang dari mutasi ini pada fungsi sel rambut dan kelangsungan hidup pada model hewan akan sangat berharga dalam memahami sifat progresif DFNA15 dan dalam mengembangkan intervensi potensial untuk memperlambat atau mencegah perkembangan gangguan pendengaran.

