
Abstrak
Bubuk yang aman bagi anak-anak untuk suspensi oral (PfOS) tovorafenib, inhibitor RAF tipe II, dikembangkan untuk pasien yang mengalami kesulitan menelan tablet. Studi fase 1 acak berlabel terbuka (QSC205140) ini mengevaluasi rasa/kenikmatan formulasi PfOS (n = 12), bioavailabilitas relatif PfOS versus formulasi tablet, dan efek makanan pada tablet (n = 12) pada peserta yang sehat. Tovorafenib awalnya diberikan sebanyak 300 mg dan dikurangi menjadi 100 mg karena efek samping muskuloskeletal (AE). Penambahan pemanis dan/atau perasa meningkatkan rasa/kenikmatan. Rasio rata-rata geometris (interval kepercayaan 90%) dari konsentrasi obat plasma puncak yang dikoreksi dosis (Cmax / D) dan area di bawah kurva konsentrasi plasma-waktu dari waktu nol hingga konsentrasi terukur terakhir (AUC 0-terakhir /D) antara formulasi PfOS dan tablet masing-masing adalah 96% (83%-111%) dan 104% (95%-115%). Dibandingkan dengan pemberian saat puasa, pemberian tablet dengan makanan mengakibatkan penundaan waktu hingga Cmax selama 2-3,5 jam , dan penurunan Cmax /D sebesar 20% tanpa perubahan pada AUC 0-terakhir /D. Empat AE parah dan 7 AE sedang terjadi dengan 300 mg tovorafenib. Semua AE yang tersisa, yang dilaporkan dengan 100 mg dan 300 mg, bersifat ringan. Data ini menunjukkan bahwa PfOS tovorafenib dan formulasi tablet sebanding, dan bahwa tablet dapat diberikan dengan atau tanpa makanan.
Glioma tingkat rendah pediatrik (pLGG) merupakan tumor sistem saraf pusat anak yang paling umum dan mencakup sekitar 30% dari tumor otak anak, dengan sekitar 1000-1600 kasus baru yang diantisipasi terjadi setiap tahunnya di Amerika Serikat. 1 – 3 Perubahan pada homolog onkogen virus sarkoma murine v-Raf ( BRAF ) terdapat pada hingga 70% kasus pLGG. 4 , 5
Tovorafenib adalah inhibitor RAF tipe II, molekul kecil, selektif, dan penetrasi sistem saraf pusat yang diberikan secara oral dengan aktivitas melawan bentuk monomerik (perubahan Kelas I) dan dimerik (perubahan Kelas II, termasuk fusi) dari sinyal RAF. 6 Tovorafenib juga dikenal sebagai AMG-2112819, BIIB-024, BSK1369, MLN-2480, TAK-580, dan DAY101, dan nama kimianya adalah 6-amino-5-kloro-N-[(1R)-1-[5-[[[5kloro-4-(trifluorometil)-2-piridinil]amino]karbonil]-2-tiazolil]etil]-4-pirimidinkarboksamida. 6 , 7 Dalam beberapa studi klinis, tovorafenib menunjukkan bukti aktivitas antitumor pada pasien dengan tumor yang memiliki fusi BRAF atau mutasi BRAF V600E. 6 , 8-12 Tovorafenib digunakan untuk pengobatan pasien berusia 6 bulan ke atas dengan pLGG kambuh atau refrakter yang mengandung fusi atau penataan ulang BRAF , atau mutasi BRAF V600. 13 Pada 76 pasien yang dapat dievaluasi dengan pLGG kambuh/refrakter pada kelompok FIREFLY-1 1 (NCT04775485), tovorafenib (420 mg/m 2 sekali seminggu ; maksimum 600 mg) menunjukkan tingkat respons keseluruhan sebesar 51% oleh komite peninjau radiologi independen berdasarkan kriteria glioma tingkat rendah pada Penilaian Respons pada Neuro-Onkologi Pediatrik. Durasi median respons adalah 13,8 bulan. 13 , 14 Uji coba Fase 3 LOGGIC/FIREFLY-2 (NCT05566795) yang mengevaluasi tovorafenib (380 mg/m 2 sekali seminggu; maksimum 600 mg) pada pLGG lini pertama yang diubah BRAF dibandingkan dengan kemoterapi perawatan standar sedang dilakukan secara global. 15
Farmakokinetik (PK) tovorafenib telah dipelajari pada pasien dewasa dengan tumor padat lanjut. 12 Konsentrasi plasma puncak dan paparan tovorafenib kira-kira proporsional dengan dosis pada rentang dosis 20-280 mg setiap 2 hari dan 400-800 mg setiap minggu, dengan waktu paruh sekitar 70 jam (rentang 31-119 jam), dan waktu rata-rata hingga konsentrasi plasma maksimum yang teramati (tmax ) 3 jam (rentang 1-24 jam) setelah pemberian dosis. 12 Studi in vitro menunjukkan bahwa tovorafenib terutama dimetabolisme oleh aldehida oksidase dan sitokrom P450 (CYP) 2C8, dengan kontribusi kecil dari CYP3A, CYP2C9, dan CYP2C19. 6 , 16 Sebuah studi Fase 1 [ 14 C]-tovorafenib pada dosis oral tunggal 100 mg pada partisipan pria sehat menunjukkan bahwa tovorafenib adalah komponen sirkulasi yang paling melimpah dalam plasma, mencakup 78,7% dari total paparan radioaktivitas plasma. Setelah dosis oral tunggal tovorafenib berlabel radioaktif, 65% dari total dosis berlabel radioaktif ditemukan dalam feses (8,6% tidak berubah) dan 27% dari dosis ditemukan dalam urin (0,2% tidak berubah), yang menunjukkan bahwa rute utama eliminasi adalah metabolisme hati dan ekskresi bilier. 6 , 16 Studi in vitro telah menunjukkan bahwa tovorafenib menghambat dan menginduksi enzim CYP tertentu, termasuk CYP2C8, CYP2C9, CYP2C19, dan CYP3A pada konsentrasi yang relevan secara klinis. 6 , 16 Tovorafenib juga menginduksi CYP1A2 dan CYP2B6 dan menghambat protein resistensi kanker payudara pada konsentrasi yang relevan secara klinis. 6 , 16 Studi klinis interaksi obat-obat terkait tovorafenib sedang berlangsung.
Tovorafenib awalnya diformulasikan sebagai tablet untuk uji klinis pada orang dewasa. Namun, karena pendaftaran dalam uji klinis FIREFLY-1 dilakukan pada populasi anak-anak (usia 6 bulan hingga 25 tahun), penting untuk mengembangkan formulasi bubuk tovorafenib untuk suspensi oral (PfOS) guna menyediakan formulasi alternatif yang lebih mudah diberikan bagi peserta yang tidak dapat menelan tablet. Lebih jauh, PfOS juga dapat memberikan dosis yang lebih akurat berdasarkan luas permukaan tubuh untuk pasien yang lebih muda dibandingkan dengan pemberian formulasi tablet.
Studi Fase 1 acak, berlabel terbuka, 2 bagian, satu pusat (QSC205140) ini bertujuan untuk mengevaluasi dan membandingkan PfOS baru tovorafenib dengan tablet yang ada.
Metode
Desain Studi
Bagian 1 dari penelitian ini mengevaluasi atribut rasa dan palatabilitas (bau, rasa manis, rasa pahit, rasa, tekstur/rasa di mulut, dan aftertaste) dan penerimaan keseluruhan tovorafenib PfOS dengan pemanis dan rasa tambahan. Formulasi dengan tingkat rasa dan pemanis yang optimal digunakan untuk evaluasi lebih lanjut di Bagian 2 dari penelitian ini. Rasa dan palatabilitas di Bagian 2 dari penelitian ini menentukan bioavailabilitas relatif dan mengevaluasi profil PK dari tovorafenib PfOS yang disukai dari Bagian 1 dibandingkan dengan tablet dan menilai efek potensial makanan pada PK tovorafenib setelah dosis tunggal tablet pada peserta sehat dalam keadaan kenyang menggunakan desain crossover 3 periode acak. Penelitian ini dilakukan oleh Quotient Sciences–Miami, Inc. (Miami, FL) atas nama Day One Biopharmaceuticals, Inc. Protokol penelitian dan formulir persetujuan telah disetujui oleh Institutional Review Board (Advarra, Columbia, MD). Semua peserta secara sukarela menandatangani formulir persetujuan sebelum prosedur khusus penelitian dilakukan.
Peserta
Peserta pria dan wanita sehat berusia antara 18 dan 55 tahun dengan indeks massa tubuh 18,0-32,0 kg/m 2 diikutsertakan dalam penelitian ini. Peserta wanita yang memenuhi syarat termasuk mereka yang tidak hamil atau menyusui di Bagian 1 penelitian dan yang tidak berpotensi untuk melahirkan di Bagian 2 penelitian. Kriteria eksklusi utama termasuk kimia klinis, hematologi, atau urinalisis abnormal yang signifikan secara klinis; riwayat penyakit kardiovaskular, ginjal, hati, pernapasan kronis, atau gastrointestinal yang signifikan secara klinis (kecuali kolesistektomi); gangguan neurologis atau psikiatris, sebagaimana dinilai oleh peneliti; penyakit kulit serius yang signifikan, termasuk ruam, alergi makanan, eksim, psoriasis, atau urtikaria; dan peserta yang sedang mengonsumsi, atau telah mengonsumsi, obat apa pun yang diresepkan atau yang dijual bebas atau pengobatan herbal (selain hingga 4 g/hari asetaminofen atau terapi penggantian hormon; kontrasepsi hormonal diizinkan di Bagian 1 saja) dalam 14 hari sebelum pemberian tovorafenib.
Penilaian Rasa dan Palatabilitas
Pada Bagian 1 penelitian, Formulasi A merupakan acuan dan tidak mengandung pemanis atau perasa, Formulasi B hanya mengandung pemanis (sukralosa), dan Formulasi C hingga F mengandung perasa stroberi atau vanila dengan kadar pemanis (sukralosa) yang berbeda-beda (Tabel 1 ). Semua formulasi dilarutkan kembali menjadi 25 mg/mL tovorafenib.
Tabel 1. Ringkasan Formulasi PfOS

PfOS, bubuk untuk suspensi oral.
Dua belas peserta menerima pelatihan dalam teknik “sip and spit” pada hari sebelum pemberian salah satu formulasi PfOS dan menyelesaikan kuesioner praktik. Pada hari penelitian, peserta diacak ke 1 dari 6 urutan (ABFCED, BCADFE, CDBEAF, DECFBA, EFDACB, dan FAEBDC, dengan 2 peserta ditugaskan untuk setiap urutan) (Gambar S1A ). Peserta menerima 5 mL PfOS tertentu dan diinstruksikan untuk menahannya di mulut mereka selama sekitar 30 detik sebelum ekspektorasi. Peserta diinstruksikan untuk tidak menelan PfOS apa pun. Ada pencucian minimal 1 jam di antara mencicipi setiap formulasi. Selama waktu ini, peserta membersihkan langit-langit mulut mereka menggunakan air mineral dan kerupuk tawar. Rasa dan palatabilitas dinilai menggunakan kuesioner di mana partisipan menilai penerimaan keseluruhan setiap formulasi (terlepas dari formulasi sebelumnya) pada atribut palatabilitas tertentu (bau, kemanisan, kepahitan, rasa, rasa/tekstur di mulut, dan sisa rasa) pada skala 9 poin (1 = sangat tidak suka; 2 = sangat tidak suka; 3 = agak tidak suka; 4 = agak tidak suka; 5 = netral maupun suka; 6 = agak suka; 7 = agak suka; 8 = sangat suka; atau 9 = sangat suka) terlepas dari formulasi sebelumnya. Preferensi untuk formulasi rasa tertentu dikelompokkan sebagai berikut: Tingkat 1-3 = tidak suka; Tingkat 4-6 = netral; Tingkat 7-9 = suka. Skala hedonik 9 poin ini dikembangkan dan divalidasi oleh Peryam et al. 17 sebagai metode pengukuran preferensi makanan dan telah digunakan dalam penilaian rasa/palatabilitas untuk produk farmasi. 18 , 19 Skala penilaian yang digunakan disediakan dalam Gambar S2 . Setiap peserta memiliki waktu hingga 15 menit untuk menyelesaikan kuesioner secara individu dan pribadi sebelum mencicipi PfOS berikutnya.
Bioavailabilitas Relatif dan Penilaian Efek Pangan
Bahasa Indonesia: Pada Bagian 2 penelitian, 12 partisipan diacak ke dalam 1 dari 3 urutan (T[puasa]S[puasa]T[makan], S[puasa]T[makan]T[puasa], dan T[makan]T[puasa]S[puasa]) (Gambar S1B ). Pada setiap periode pengobatan, tovorafenib diberikan pada Hari 1 sebagai tablet saat berpuasa (T[puasa]), sebagai PfOS saat berpuasa (S[puasa]), atau sebagai tablet setelah makan berlemak tinggi (T[makan]). Tovorafenib diberikan pada pagi hari setelah puasa semalaman selama 10 jam atau lebih. Untuk kelompok pengobatan T[puasa] dan S[puasa], tovorafenib diberikan dalam kondisi berpuasa (yaitu, tanpa sarapan), sedangkan untuk kelompok T[makan], diberikan 30 menit setelah dimulainya makan berlemak tinggi. Sebanyak 240 mL air diberikan dengan setiap tablet tovorafenib. Untuk PfOS, segera setelah pemberian, wadah dosis dibilas dengan air, dan peserta diminta untuk mengonsumsi larutan pembilas dan air tambahan hingga volume total 240 mL (termasuk volume dosis dan volume yang digunakan untuk membilas wadah dosis). Jika diperlukan, air tambahan dalam alikuot 50 mL disediakan dan dicatat tetapi tidak digolongkan sebagai penyimpangan protokol. Peserta diminta untuk berpuasa selama 4 jam atau lebih setelah pemberian dosis tunggal tablet tovorafenib. Komposisi makanan berlemak tinggi terdiri dari 800-1000 kalori, yang sekitar 54% kalorinya berasal dari lemak. Agar memenuhi syarat untuk pemberian dosis, peserta diminta untuk mengonsumsi setidaknya 90% dari makanan yang disediakan. Asupan cairan dilarang 1 jam sebelum pemberian dosis hingga 1 jam setelah pemberian dosis, kecuali cairan yang dikonsumsi dengan sarapan sebelum pemberian dosis dan air untuk asupan tablet tovorafenib. Sampel darah dikumpulkan secara berkala (lihat Pengumpulan dan Pemrosesan Darah untuk Analisis PK di bagian Penilaian Farmakokinetik untuk keterangan lebih rinci) untuk analisis PK dari sebelum pemberian dosis hingga 120 jam setelah pemberian dosis. Peserta diminta untuk tetap berada di unit klinis hingga 96 jam setelah pemberian dosis (Hari ke-5) dan kembali ke klinik pada 120 jam setelah pemberian dosis (Hari ke-6) sehingga sampel darah terakhir dapat diambil untuk analisis PK. Ada jeda minimal 14 hari di antara setiap dosis. Panggilan tindak lanjut dilakukan 7-10 hari setelah dosis terakhir untuk memastikan kesejahteraan peserta yang berkelanjutan. Peserta dapat menarik diri dari penelitian kapan saja dengan alasan apa pun.
Pada Periode 1, peserta menerima 300 mg tovorafenib dalam bentuk tablet (3 x 100 mg tablet tovorafenib) atau PfOS dalam keadaan berpuasa, atau hanya satu tablet setelah makan makanan berlemak tinggi. Kekuatan dosis tertinggi untuk tablet tovorafenib adalah 100 mg. Ke-12 peserta mengalami efek samping muskuloskeletal (AE) yang tidak serius setelah pemberian tovorafenib pada dosis ini; dengan demikian, untuk Periode 2 dan 3, dosis dikurangi menjadi 100 mg. Peserta juga diberi premedikasi dengan ibuprofen untuk mengurangi AE muskuloskeletal. Dosis awal 400 mg ibuprofen diberikan antara 30 menit dan 1 jam sebelum pemberian dosis tovorafenib dan kemudian kira-kira setiap 8 jam (± 1 jam) setelah pemberian dosis, hingga 48 jam setelah pemberian dosis. Setelah 48 jam, peserta dapat terus mengonsumsi ibuprofen sesuai kebutuhan.
Penilaian Farmakokinetik
Pengumpulan dan Pemrosesan Darah untuk Analisis PK
Selama setiap periode pengobatan, sampel darah serial dikumpulkan sebelum pemberian dosis dan hingga 120 jam setelah pemberian dosis pada titik waktu berikut untuk penentuan konsentrasi plasma tovorafenib: 0 jam sebelum pemberian dosis dan 0,5, 1, 1,5, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24, 40, 48, 60, 72, 96, dan 120 jam setelah pemberian dosis. Sampel darah dikumpulkan dalam tabung asam dipotassium etilendiamintetraasetat dan disentrifugasi dalam waktu 30 menit setelah pengumpulan. Setelah sentrifugasi, sebagian plasma yang dipanen dipindahkan ke dalam krioval dan disimpan pada suhu -70°C hingga tiba saatnya untuk analisis sampel.
Konsentrasi plasma tovorafenib ditentukan dengan kromatografi cair tervalidasi dengan metode berbasis spektrometri massa tandem dengan batas kuantifikasi bawah 0,5 ng/mL.
Setelah sampel dicairkan, alikuot plasma sebanyak 50 µL ditambahkan ke pelat 96-sumur, diikuti dengan penambahan 50 µL larutan standar internal berlabel isotop (5x 13 C-tovorafenib). Protein plasma diendapkan dengan penambahan 300 µL larutan asetonitril:asam format pada rasio 100:1 (v:v). Pelat ditutup, diaduk, dan disentrifugasi selama kurang lebih 10 menit pada kecepatan 3000 rpm sebelum pemindahan sekitar 300 µL supernatan ke pelat 96-sumur bersih lainnya. Supernatan dikeringkan seluruhnya, kemudian 200 µL pelarut rekonstitusi (asetonitril:air pada 50:50 [v:v]) ditambahkan ke dalam pelat 96 sumur dan dicampur lagi dengan vortex sebelum sampel diinjeksikan ke dalam kromatografi cair kinerja tinggi fase terbalik dengan sistem spektrometri massa tandem Turbo Ion Spray.
Metode gradien fase terbalik yang berjalan pada laju alir 0,40 mL/menit pada kolom Venusil C18 5 µm, 2,1 × 50 mm (Phenomenex) menghasilkan waktu retensi tovorafenib sekitar 1,70 menit. Fase bergerak yang digunakan adalah air:asam format pada 100:0,1 (v:v) (A); dan asetonitril:air:asam format pada 95:5:0,1 (v:v:v) (B) di bawah mode semprotan ion positif dan dideteksi melalui pemantauan reaksi ganda pasangan transisi massa pada m / z 506,2→334,2 dan 511,2→334,2 untuk tovorafenib dan standar internalnya, masing-masing.
Kurva kalibrasi dibuat menggunakan rasio luas puncak analit terhadap standar internal berlabel isotopik. Linearitas dengan regresi kuadrat terkecil tertimbang 1/x2 dicapai dalam rentang konsentrasi tovorafenib 0,500-3500 ng/mL dengan sampel kontrol kualitas berkisar antara 1,5 hingga 2800,0 ng/mL. Hasil untuk presisi dan akurasi intraday masing-masing adalah 0,6%-12,6% dan -4,4% hingga 3,6%; presisi dan akurasi interday masing-masing adalah 1,9%-9,5% dan -1,3 hingga 1,4%. Analisis ulang sampel yang terjadi dilakukan pada sekitar 10% sampel studi, dan hasilnya memenuhi kriteria penerimaan.
Analisis Farmakokinetik
Parameter PK yang dinilai meliputi area di bawah kurva konsentrasi plasma–waktu (AUC) dari waktu nol hingga waktu konsentrasi terakhir yang dapat diukur (AUC 0–terakhir ), AUC dari waktu nol hingga tak terhingga (AUC 0–inf ), konsentrasi plasma maksimum yang teramati setelah pemberian obat (C max ), waktu hingga C max (t max ), dan waktu paruh eliminasi yang dikaitkan dengan kemiringan terminal kurva konsentrasi–waktu semilogaritmik (t 1/2 ).
Penilaian Keamanan
Evaluasi keamanan mencakup analisis AE, variabel laboratorium (hematologi, kimia klinis, dan urinalisis), tanda-tanda vital, elektrokardiogram, dan pemeriksaan fisik. AE dikodekan menggunakan Kamus Medis untuk Aktivitas Regulasi Versi 24.0. Tingkat keparahan AE dinilai sebagai berikut: “ringan” didefinisikan sebagai AE yang mudah ditoleransi oleh peserta, menyebabkan ketidaknyamanan minimal, dan tidak mengganggu aktivitas sehari-hari; “sedang” didefinisikan sebagai AE yang cukup tidak nyaman untuk mengganggu aktivitas sehari-hari yang normal, dan intervensi mungkin diperlukan; dan “berat” didefinisikan sebagai AE yang mencegah aktivitas sehari-hari yang normal dan pengobatan, atau intervensi lain, diperlukan. AE yang muncul akibat pengobatan (TEAE) dirangkum berdasarkan frekuensi, tingkat keparahan, dan keterkaitan dengan obat studi. Frekuensi TEAE (jumlah TEAE dan jumlah peserta yang mengalami TEAE) ditabulasikan berdasarkan kelas organ sistem dan istilah yang disukai. Dalam analisis per peserta, peserta yang mengalami kejadian yang sama lebih dari satu kali hanya dihitung satu kali. Reaksi obat yang merugikan didefinisikan sebagai AE apa pun yang hubungan kausalnya dengan obat yang diteliti setidaknya merupakan kemungkinan yang wajar, yaitu, “berhubungan.” AE serius dianggap sebagai AE apa pun yang mengakibatkan kematian, mengancam jiwa atau memerlukan rawat inap, mengakibatkan kecacatan/ketidakmampuan yang terus-menerus atau signifikan, atau merupakan kejadian medis penting sebagaimana diakui oleh peneliti.
Analisis Statistik
Untuk Bagian 1 dari penelitian ini, peserta yang dapat dievaluasi didefinisikan sebagai peserta yang telah menyelesaikan kuesioner rasa/palatabilitas untuk semua formulasi. Setiap atribut rasa/palatabilitas dan penerimaan keseluruhan dibandingkan di antara 6 formulasi menggunakan uji Friedman nonparametrik pada tingkat signifikansi 5%. Hipotesis nolnya adalah tidak ada perbedaan yang signifikan secara statistik antara formulasi dalam setiap atribut rasa/palatabilitas atau penerimaan keseluruhan. Jika uji Friedman menunjukkan signifikansi pada tingkat 5%, perbandingan berpasangan dilakukan menggunakan uji peringkat bertanda Wilcoxon, dan nilai P ditetapkan untuk setiap formulasi uji (Formulasi B hingga F) versus referensi (Formulasi A) untuk setiap atribut dan penerimaan keseluruhan. Karena sifat penelitian yang eksploratif, tidak ada penyesuaian untuk beberapa perbandingan, dan nilai P disajikan hanya untuk tujuan deskriptif.
Untuk Bagian 2 dari penelitian ini, parameter PK plasma untuk tovorafenib dihitung dari profil konsentrasi plasma individu dengan metode analisis nonkompartemen, menerapkan metode trapesium linear, yang diterapkan dalam Phoenix WinNonlin (Versi 8.0, Certara USA, Inc.). Statistik deskriptif (rata-rata, deviasi standar, rasio rata-rata geometrik [GMR], dan koefisien variasi geometrik) dihitung untuk semua parameter PK kecuali untuk t max , yang hanya median dan rentangnya yang ditentukan. Model efek campuran linear dipasangkan pada parameter PK yang ditransformasikan log (AUC 0–last , AUC 0–inf , dan C max ). Model tersebut mencakup pengobatan, periode, dan urutan sebagai faktor tetap dan peserta yang disarangkan dalam urutan sebagai faktor acak. Analisis statistik formal dilakukan pada parameter PK yang dikoreksi dosis C max , AUC 0–last , dan AUC 0–inf untuk menilai bioavailabilitas relatif dan keberadaan efek makanan pada tovorafenib. Untuk mengevaluasi bioavailabilitas relatif tovorafenib PfOS (puasa) dibandingkan dengan tablet tovorafenib (puasa), dan efek makanan pada tablet tovorafenib (makan dibandingkan dengan puasa), estimasi titik dan interval kepercayaan 90% yang sesuai untuk perbedaan antara rata-rata kuadrat terkecil dari uji (PfOS, puasa; atau tablet, makan) dan referensi (tablet, puasa) dihitung. Ini ditransformasikan kembali untuk mendapatkan estimasi titik dan interval kepercayaan 90% untuk GMR pada skala yang tidak ditransformasi. Set analisis PK, yang ditetapkan untuk Bagian 2 saja, mencakup semua peserta yang menerima setidaknya 1 dosis obat studi dan yang memenuhi kriteria berikut untuk setidaknya 1 profil: tidak ada sampel yang hilang atau hasil analitis pascadosis yang tidak valid pada titik waktu kritis (misalnya, sekitar Cmax ) ; tidak ada penyimpangan protokol relevan yang dapat memengaruhi tujuan studi sehubungan dengan titik akhir PK; tidak ada AE relevan seperti muntah yang menunjukkan bahwa seluruh dosis tidak tersedia untuk penyerapan untuk peserta tertentu. Subset analisis PK, yang ditetapkan hanya untuk Bagian 2, mencakup partisipan yang dapat dievaluasi yang menerima regimen T[puasa] dan S[puasa] dan yang memiliki data PK hingga 72 jam setelah pemberian dosis untuk penilaian bioavailabilitas relatif, dan partisipan yang dapat dievaluasi yang telah menerima regimen T[puasa] dan T[makan] dan yang memiliki data PK hingga 72 jam setelah pemberian dosis untuk penilaian efek makanan. Parameter PK dikecualikan dari statistik ringkasan jika nilai pradosis yang terukur diamati lebih besar dari 5% dari C maks .
Nilai-nilai yang tidak terukur yang dilaporkan dalam data konsentrasi plasma (yaitu, nilai-nilai yang berada di bawah batas kuantisasi [BLQ]) ditetapkan ke nol untuk penentuan statistik ringkasan, kecuali untuk rata-rata geometrik, deviasi standar geometrik, dan koefisien variasi geometrik, di mana nilai-nilai BLQ diperhitungkan sebagai setengah dari batas bawah nilai kuantifikasi. Untuk semua plot pada skala linier, nilai-nilai konsentrasi yang dilaporkan sebagai BLQ disajikan sebagai nol.
Tidak ada perhitungan ukuran sampel formal yang dilakukan, karena penelitian ini bersifat eksploratif. Dua belas partisipan didaftarkan untuk setiap bagian penelitian guna memastikan minimal 10 partisipan yang dapat dievaluasi, yang dianggap tepat untuk memenuhi tujuan penelitian.
Hasil
Populasi Studi
Pada Bagian 1 penelitian, 12 peserta diacak ke 1 dari 6 sekuens dan menyelesaikan penelitian. Semua 12 peserta menerima sedikitnya 1 dosis tovorafenib dan dimasukkan dalam set analisis keamanan dan rasa. Pada Bagian 2 penelitian, 12 peserta didaftarkan dan diacak ke 1 dari 3 sekuens. Semua 12 peserta menerima sedikitnya 1 dosis tovorafenib, memiliki minimal 1 hasil analitis pascadosis yang valid untuk estimasi parameter PK, dan memenuhi kriteria untuk sedikitnya 1 profil. Oleh karena itu, semua 12 peserta dimasukkan dalam set analisis keamanan dan PK. Satu peserta tidak menerima formulasi referensi (T[cepat]) dan oleh karena itu dikeluarkan dari subset analisis PK untuk analisis statistik formal. Ringkasan peserta dan karakteristik dasar disajikan dalam Tabel S1 . Pada Bagian 1 dan 2, mayoritas peserta berkulit putih (masing-masing 91,7% dan 83,3%), dengan distribusi wanita dan pria yang sama. Usia rata-rata adalah 40,3 (kisaran, 36-52) tahun di Bagian 1 dan 42,5 (kisaran, 35-54) tahun di Bagian 2. Indeks massa tubuh rata-rata adalah 26,4 kg/m2 di Bagian 1 dan 27,6 kg/m2 di Bagian 2.
Evaluasi Atribut Rasa dan Palatabilitas
Skor median untuk setiap atribut rasa/palatabilitas (misalnya, bau, kemanisan, kepahitan, rasa, tekstur/rasa di mulut, dan sisa rasa) dan penerimaan keseluruhan dirangkum dalam Tabel 2. Skor median terendah, 3,0, diamati dalam formulasi referensi (Formulasi A) untuk rasa, tekstur/rasa di mulut, sisa rasa, dan penerimaan keseluruhan. Penambahan pemanis (Formulasi B) meningkatkan skor median untuk semua 3 atribut ini, serta penerimaan keseluruhan. Perasa yang dipasangkan dengan jumlah pemanis yang berbeda (Formulasi C hingga F) memberikan peningkatan yang lebih kuat atas produk referensi untuk penerimaan keseluruhan dan setiap atribut rasa kecuali kepahitan, seperti yang ditunjukkan oleh skor median yang lebih meningkat. Tampaknya tidak ada preferensi yang jelas untuk formulasi rasa tertentu. Formulasi dengan proporsi tertinggi yang dinilai sebagai “suka” adalah Formulasi C (0,4% perasa stroberi dan 0,15% sukralosa).
Tabel 2. Skor Atribut Rasa/Palatabilitas dan Penilaian untuk Formulasi Tovorafenib PfOS (Perlakuan Uji 0,4% Perasa/Sukralosa%)
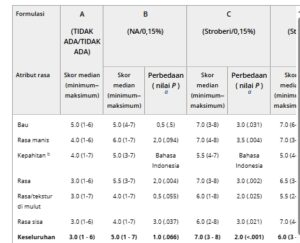
NA, tidak berlaku; PfOS, bubuk untuk suspensi oral.
Kunci untuk nilai skor median: 1 = sangat tidak suka, 2 = sangat tidak suka, 3 = agak tidak suka, 4 = agak tidak suka, 5 = netral, 6 = agak suka, 7 = cukup suka, 8 = sangat suka, 9 = sangat suka. Peserta mencicipi 5 mL dari 6 formulasi PfOS yang berbeda (A–F). Formulasi A = 25 mg/mL tovorafenib tanpa pemanis/perasa; Formulasi B hingga F = 25 mg/mL tovorafenib dengan berbagai tingkat perasa dan sukralosa.
Median dari perbedaan berpasangan antara formulasi uji versus formulasi referensi A, dan nilai P dari uji peringkat bertanda Wilcoxon. Karena sifat eksploratif dari penelitian ini, tidak ada penyesuaian untuk beberapa perbandingan, dan nilai P bersifat deskriptif.
b Untuk atribut kepahitan, nilai P >0,05 dari uji Friedman; oleh karena itu, tidak dilakukan uji peringkat bertanda Wilcoxon.
Uji Friedman secara statistik signifikan untuk penerimaan keseluruhan dan aspek rasa seperti bau, kemanisan, rasa, rasa/tekstur di mulut, dan sisa rasa, yang menunjukkan bahwa setidaknya 1 formulasi memiliki skor rasa yang berbeda secara signifikan dari formulasi lainnya ( P < .05). Untuk kepahitan, nilai P lebih besar dari .05, yang menunjukkan tidak ada perbedaan signifikan antara setidaknya 2 regimen untuk skor kepahitan. Perbandingan berpasangan selanjutnya dilakukan dengan menggunakan uji peringkat bertanda Wilcoxon (Tabel 2 ). Untuk semua perbandingan berpasangan, median perbedaan berpasangan adalah positif, yang menunjukkan bahwa setiap kombinasi rasa/pemanis menunjukkan peningkatan penerimaan untuk setiap aspek rasa yang dianalisis dan penerimaan keseluruhan jika dibandingkan dengan Formulasi referensi A tanpa pemanis atau perasa. Peningkatan terkecil dari Formulasi A diamati dengan Formulasi B (sukralosa 0,15%) di seluruh aspek rasa, kecuali untuk sisa rasa, di mana peningkatannya sama dengan regimen dengan perasa. Formulasi C (rasa stroberi dan sukralosa 0,15%), Formulasi D (rasa stroberi dan sukralosa 0,3%), dan Formulasi E (rasa vanila dan sukralosa 0,3%), semuanya menunjukkan peningkatan serupa di seluruh aspek rasa dan penerimaan keseluruhan jika dibandingkan dengan Formulasi A acuan, dengan peningkatan ini secara umum lebih besar daripada Formulasi F (rasa vanila dan sukralosa 0,15%).
Secara keseluruhan, skor rasa meningkat secara signifikan untuk formulasi tovorafenib PfOS dengan penambahan pemanis (sukralosa) saja atau dengan tambahan rasa stroberi atau vanila, dengan peningkatan yang lebih besar terlihat dengan penambahan pemanis dan rasa untuk penerimaan keseluruhan dan semua aspek rasa. Formulasi C (0,4% rasa stroberi dan 0,15% sukralosa) dipilih untuk penilaian bioavailabilitas relatif di Bagian 2 berdasarkan hasil rasa dan palatabilitas keseluruhan dari Bagian 1.
Bioavailabilitas Relatif dan Efek Makanan
Profil konsentrasi plasma rata-rata–waktu tovorafenib di seluruh 3 regimen (T[puasa], S[puasa], dan T[makan]) pada dosis 100 mg dan 300 mg masing-masing disajikan pada Gambar 1A , 1B .
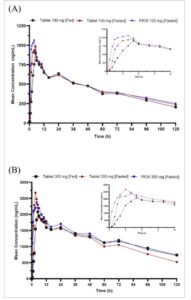
Gambar 1
Buka di penampil gambar
Kekuatan Gambar
Profil konsentrasi plasma rata-rata–waktu untuk tovorafenib pada dosis 100 mg dan 300 mg dalam berbagai kondisi dan formulasi prandial (tablet vs. PfOS). Nilai di bawah batas kuantifikasi diberi nilai nol dan dimasukkan dalam perhitungan nilai rata-rata. (A) Tovorafenib 100 mg dan (B) tovorafenib 300 mg. PfOS, bubuk untuk suspensi oral.
Ringkasan parameter PK dengan dosis tunggal 100 atau 300 mg tovorafenib di seluruh 3 regimen disediakan dalam Tabel 3. Dalam kondisi berpuasa, paparan serupa (AUC 0–terakhir dan Cmax ) diamati dengan formulasi tablet dan PfOS pada dosis 100 dan 300 mg. Pemberian tablet tovorafenib dengan makanan berlemak tinggi menyebabkan penyerapan tertunda, di mana median t max 1,5 dan 3,5 jam lebih lama dibandingkan dengan kondisi berpuasa pada 100 dan 300 mg, masing-masing. Selain itu, nilai C max sedikit lebih tinggi dalam kondisi berpuasa daripada diberi makan. Namun, keseluruhan paparan AUC 0–terakhir serupa antara keadaan berpuasa dan diberi makan, yang menunjukkan makanan tidak memengaruhi tingkat penyerapan.
Tabel 3. Ringkasan Parameter PK Dengan 1 Dosis Tovorafenib 100 dan 300 mg
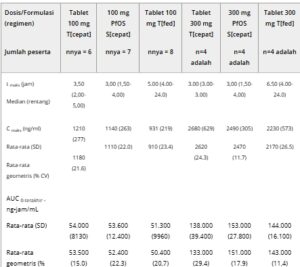
AUC 0–terakhir , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga waktu konsentrasi terukur terakhir; C max , konsentrasi plasma maksimum yang teramati; CV, koefisien variasi; PfOS, serbuk untuk suspensi oral; PK, farmakokinetik; SD, deviasi standar; S[puasa], PfOS saat berpuasa; t 1/2 , waktu paruh eliminasi yang dikaitkan dengan kemiringan terminal kurva konsentrasi–waktu semilogaritmik; t max , waktu hingga konsentrasi plasma maksimum yang teramati; T[puasa], tablet saat berpuasa; T[makan], tablet setelah makan berlemak tinggi.
Parameter PK tidak dimasukkan dalam statistik ringkasan karena R 2 yang disesuaikan sebesar <0,9. Parameter PK rata-rata (aritmatika dan geometrik) untuk plasma tovorafenib setelah dosis oral tunggal (100 mg dan 300 mg) diberikan kepada peserta studi dalam bentuk tablet atau formulasi PfOS di Bagian 2 studi.
Dalam studi terkini, Cmax dan AUC 0–last pada 300 mg kira-kira 2,3 dan 2,5 kali lipat dari yang diamati pada 100 mg, masing-masing. Dengan demikian, perbandingan statistik parameter PK tovorafenib yang dikoreksi dosis (Cmax dan AUC 0–last ) selanjutnya dilakukan untuk menilai bioavailabilitas relatif dan efek makanan. Ringkasan GMR Cmax / Dosis (D) dan AUC 0–last /D antara uji PfOS (S[puasa]) dan formulasi referensi tablet (T[puasa]), bersama dengan parameter paparan sistemik dalam kondisi makan dan berpuasa, disediakan dalam Tabel 4 .
Tabel 4. Parameter PK yang Dikoreksi Dosis untuk Penilaian Bioavailabilitas Relatif dan Efek Makanan Tovorafenib
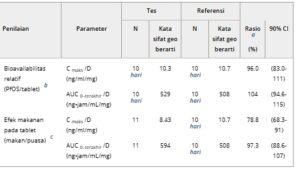
Adj. geo mean, mean geometrik yang disesuaikan dari model di mana parameter farmakokinetik (yaitu, C max dan AUC) disesuaikan dengan tingkat dosis; AUC 0–terakhir , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga waktu konsentrasi terakhir yang dapat diukur; CI, interval kepercayaan; C max , konsentrasi plasma maksimum yang diamati; D, dosis; PfOS, bubuk untuk suspensi oral; PK, farmakokinetik.
Rasio rata-rata geometrik yang disesuaikan untuk pengujian: referensi.
b PfOS: formulasi PfOS tovorafenib, puasa (uji); formulasi tablet tovorafenib, puasa (referensi).
c Makan: formulasi tablet tovorafenib (uji); berpuasa: formulasi tablet tovorafenib (referensi).
d Parameter PK dari 1 peserta dalam kelompok pengobatan dikeluarkan dari statistik ringkasan karena nilai pradosis yang terukur diamati pada >5% dari C max .
Keamanan dan Tolerabilitas
Tidak ada TEAE yang dilaporkan selama Bagian 1 penelitian. Pada Bagian 2, 12 partisipan (100%) melaporkan total 71 TEAE, 60 di antaranya terkait dengan tovorafenib (Tabel S2 ). Ada insiden reaksi obat yang merugikan yang lebih tinggi yang dilaporkan dalam regimen dosis 300 mg dibandingkan dengan regimen dosis 100 mg. Semua 4 AE berat (termasuk mialgia dan nyeri punggung) dan 7 AE sedang (termasuk mialgia, nyeri pada ekstremitas, nyeri kelenjar ludah, hipoestesia, dan parestesia) dilaporkan dalam regimen dosis 300 mg dan telah teratasi pada akhir penelitian. Semua AE dalam regimen dosis 100 mg bersifat ringan. Tidak ada AE serius atau AE yang menyebabkan penghentian obat penelitian pada Bagian 2.
Tidak ada temuan laboratorium, tanda vital, elektrokardiogram, atau pemeriksaan fisik abnormal yang signifikan secara klinis yang dicatat di kedua bagian penelitian.
Diskusi
Kebutuhan akan formulasi yang ramah bagi anak-anak telah dilaporkan secara berkala oleh para profesional perawatan kesehatan, badan regulasi, dan organisasi internasional, termasuk Organisasi Kesehatan Dunia. 20 – 22 Obat-obatan cair memungkinkan pemberian dosis berdasarkan berat badan yang lebih akurat, memudahkan penyesuaian dosis dan pemberian, serta dapat meningkatkan kepatuhan terhadap pengobatan dan, pada gilirannya, keberhasilan terapi. 22 – 24 Kebutuhan akan formulasi yang ramah bagi anak-anak ini khususnya terjadi pada pengaturan pLGG, di mana pemberian kronis sering kali diperlukan dengan terapi yang ditargetkan, dan menyoroti pentingnya studi ini yang mengevaluasi formulasi tovorafenib PfOS.
Studi ini menilai rasa dan palatabilitas dari 6 formulasi tovorafenib PfOS yang berbeda, dan Formulasi C (rasa stroberi 4% dengan sukralosa 0,15%) dianggap memiliki palatabilitas dan penerimaan keseluruhan yang paling baik. Bioavailabilitas relatif Formulasi C dibandingkan dengan tablet ditentukan. Pemberian keduanya dalam kondisi puasa menghasilkan paparan yang serupa, yang menunjukkan bahwa kedua formulasi tersebut sebanding. Secara keseluruhan, hasil tersebut mendukung tovorafenib PfOS sebagai pilihan yang layak untuk populasi anak yang menjadi sasaran.
Efek potensial makanan pada profil PK tovorafenib setelah satu dosis tablet dalam keadaan kenyang juga dinilai. Pemberian tablet tovorafenib dengan makanan berlemak tinggi menyebabkan penurunan paparan puncak sekitar 20%, tanpa memengaruhi paparan keseluruhan, yang menunjukkan bahwa tovorafenib dapat dikonsumsi dengan atau tanpa makanan. Fleksibilitas dalam mengonsumsi tovorafenib tanpa memperhatikan makanan akan memberikan kemudahan lebih bagi pasien dan pengasuh serta dapat meningkatkan kepatuhan pengobatan.
Studi klinis sebelumnya tentang tovorafenib (diberikan seminggu sekali pada 600 mg), pada pasien dengan tumor padat, menunjukkan bahwa paparan sistemik meningkat secara proporsional dosis antara 20 dan 280 mg setiap 2 hari dan 400 hingga 800 mg setiap minggu. 12 Studi ini awalnya dimulai dengan dosis tunggal 300 mg untuk mengkarakterisasi PK tovorafenib secara memadai pada peserta yang sehat. Namun, AE muskuloskeletal yang tidak serius (ringan pada 5, sedang pada 3, dan parah pada 4) diamati pada peserta yang menerima 300 mg tovorafenib, dengan mialgia sebagai AE yang paling sering dilaporkan (10 [83,3%] peserta melaporkan 10 kejadian). Dosis 300 mg dikurangi menjadi 100 mg, dan pemberian ibuprofen profilaksis diberikan kepada semua peserta dalam periode studi berikutnya. Setelah penerapan langkah-langkah ini, semua AE muskuloskeletal yang dilaporkan dalam sisa penelitian ini adalah ringan dalam tingkat keparahan. AE muskuloskeletal serupa diamati dan dilaporkan dalam studi tovorafenib dewasa dan anak Fase 1 dan Fase 2. 11 , 12 , 14 Mekanisme yang mendasari AE muskuloskeletal terkait tovorafenib belum dipelajari. Namun, AE muskuloskeletal serupa telah dilaporkan dengan inhibitor BRAF seperti encorafenib, vemurafenib, dan dabrafenib, apakah digunakan sebagai monoterapi atau dalam kombinasi dengan inhibitor mitogen-activated protein kinase, yang menunjukkan potensi efek kelas. 25 – 27 Selain itu, respons inflamasi dapat terlibat, karena subjek dengan AE muskuloskeletal nonserius dalam penelitian ini merespons dengan baik terhadap pengobatan obat antiinflamasi nonsteroid. Hal ini konsisten dengan temuan sebelumnya di mana efek samping inflamasi, termasuk artralgia dan mialgia, dari inhibitor BRAF dan MEK telah dijelaskan. 28
Nilai t 1/2 rata-rata yang diperoleh dari 3 rejimen pengobatan tovorafenib berkisar antara 62,0 hingga 89,5 jam; temuan ini serupa dengan yang dilaporkan sebelumnya pada pasien yang terpapar pada tingkat dosis yang lebih tinggi sebesar 600 mg, di mana t 1/2 rata-rata sekitar 70 jam diamati setelah beberapa kali pemberian tovorafenib. 12 Keterbatasan potensial dari penelitian saat ini mungkin adalah bahwa fase terminal yang panjang dari profil paparan tovorafenib dan jadwal pengambilan sampel menghalangi penyertaan AUC 0–inf untuk semua kelompok karena bagian kurva yang diekstrapolasi berkontribusi lebih dari 20% terhadap nilai AUC total. Perbandingan paparan keseluruhan antara formulasi, dan dalam keadaan prandial yang berbeda, dievaluasi menggunakan AUC 0–terakhir . Waktu pengambilan sampel PK terakhir adalah 120 jam setelah pemberian dosis. Meskipun tovorafenib memiliki t 1/2 yang panjang , 120 jam dianggap cukup untuk memastikan penyelesaian transit gastrointestinal (sekitar 2-3 hari) dan penyerapan tovorafenib. 29 Dengan demikian, AUC 0–last cukup untuk membandingkan tingkat penyerapan antara 2 formulasi dan untuk menilai efek makanan. Keterbatasan lain dari penelitian ini adalah bahwa penelitian ini dilakukan pada pasien dewasa yang sehat; akibatnya, temuannya mungkin tidak secara akurat mencerminkan persepsi rasa pada pasien bayi dan anak-anak, 30 – 32 terutama mereka yang telah menjalani kemoterapi. 33 Palatabilitas dan penerimaan tablet tovorafenib dan formulasi PfOS saat ini sedang dievaluasi sebagai bagian dari penelitian LOGGIC/FIREFLY-2 yang sedang berlangsung (NCT05566795) pada pLGG yang baru didiagnosis dengan BRAF yang mendaftarkan pasien anak-anak, remaja, dan dewasa muda. 15
Secara keseluruhan, hasil dari penelitian ini menunjukkan bahwa Formulasi C (rasa stroberi 4% dengan sukralosa 0,15%) dapat diterima dengan palatabilitas yang baik dan dengan paparan yang sebanding terhadap tablet. Pemberian tablet tovorafenib dengan makanan berlemak tinggi menyebabkan penurunan laju penyerapan oral; namun, hal ini tidak berdampak pada tingkat penyerapan tovorafenib secara keseluruhan, sehingga mendukung pemberian tovorafenib dengan atau tanpa makanan.

