
Abstrak
Studi Fase 1 ubrogepant ini dilakukan untuk menetapkan bioekivalensi (BE) dari formulasi tablet 50 dan 100 mg yang akan dipasarkan (TBM) dengan formulasi tablet uji klinis (CT) 100 mg dan mengevaluasi efek makanan pada bioavailabilitas tablet TBM 100 mg. Studi 2 bagian ini melibatkan peserta sehat berusia 18-45 tahun. Bagian A menilai BE setelah dosis tunggal TBM versus tablet CT. Bagian B mengevaluasi dampak makanan berlemak tinggi pada farmakokinetik ubrogepant. Keamanan dan tolerabilitas dinilai bersama dengan parameter farmakokinetik standar. Pada Bagian A (n = 47), rasio rata-rata geometrik dan interval kepercayaan 90% untuk konsentrasi obat plasma maksimum, area di bawah kurva konsentrasi plasma-waktu (AUC) dari waktu nol hingga waktu t, dan AUC dari waktu nol hingga tak terbatas untuk formulasi TBM dan CT menunjukkan BE. Waktu untuk mencapai paparan puncak sama untuk kedua formulasi. Pada Bagian B ( n = 18), makanan berlemak tinggi menunda waktu paparan puncak dan mengurangi konsentrasi obat plasma maksimum sebesar 22%, tanpa efek pada AUC. Kejadian efek samping yang muncul akibat pengobatan rendah, dan sebagian besar bersifat ringan. Tablet Ubrogepant TBM dan CT (1 × 100 mg atau 2 × 50 mg) bersifat bioekuivalen dalam kondisi puasa, dan makanan berlemak tinggi tidak memiliki efek yang relevan secara klinis pada bioavailabilitas formulasi tablet TBM.
Migrain merupakan penyebab utama kecacatan di seluruh dunia, dengan prevalensi global sebesar 14%-15%, 1 dan ditandai dengan sakit kepala berdenyut dan unilateral disertai mual, muntah, fotofobia, fonofobia, dan/atau osmofobia. 2 , 3 Perawatan akut meliputi obat-obatan yang dijual bebas (misalnya, obat antiinflamasi nonsteroid, asetaminofen) untuk mengobati sakit kepala migrain ringan hingga sedang dan triptan untuk sakit kepala migrain sedang atau berat. 2 , 3 Namun, triptan telah terbukti tidak efektif untuk sebanyak 30% pasien dengan migrain. 4 Untuk pasien ini, American Headache Society merekomendasikan penggunaan terapi yang berbeda, seperti gepan. 3
Peptida terkait gen kalsitonin (CGRP) telah lama terbukti terlibat dalam patofisiologi migrain. 5 CGRP adalah neurotransmitter yang diproduksi baik secara perifer maupun di sistem saraf pusat dan diatur oleh reseptor presinaptik yang terletak pada neuron trigeminal. 6 Selama serangan migrain, kadar CGRP meningkat, dan infus eksogen CGRP juga ditemukan dapat menyebabkan sakit kepala seperti migrain pada pasien dengan riwayat migrain, sedangkan pasien tanpa riwayat hanya mengalami sakit kepala ringan. 7 , 8 Mayoritas serat Aδ trigeminal yang menginervasi dura mengandung CGRP, 9 menunjukkan bahwa serat ini mungkin terlibat dalam peradangan neurogenik steril dan transmisi nyeri migrain. 10 Lebih jauh lagi, reseptor CGRP terdapat dalam pembuluh darah meningeal dan otak manusia. 5 , 11 Bersama-sama, pengamatan ini menunjukkan bahwa aktivasi sistem trigeminovaskular yang dimediasi CGRP mungkin memainkan peran kunci dalam patogenesis migrain dan bahwa penghambatan fungsi CGRP telah terbukti menjadi pendekatan terapeutik untuk mengobati migrain.
Ubrogepant adalah antagonis reseptor CGRP yang disetujui untuk pengobatan migrain akut pada orang dewasa. 12 Ubrogepant secara selektif mengikat reseptor CGRP dengan afinitas tinggi (K i 0,07 nM) dan memblokir sinyal siklik adenosin monofosfat yang dimediasi CGRP. 12 Dengan mencegah pengikatan CGRP, ubrogepant dianggap memblokir peradangan neurogenik yang diinduksi CGRP dan/atau menghambat penyaluran sinyal nyeri sentral dari saraf trigeminal ke nukleus trigeminal kaudatus, sehingga meredakan migrain. 12 Dalam studi Fase 1, dosis tunggal sekali sehari (1-400 mg) dan dosis ganda (hingga 400 mg) ubrogepant telah dievaluasi. 12 – 16 Dalam 2 uji coba penting Fase 3 (NCT02828020 dan NCT02867709), ubrogepant pada dosis yang disetujui memiliki kemanjuran yang lebih unggul dibandingkan plasebo dalam mencapai kebebasan dari rasa sakit 2 jam setelah pemberian dosis ( P < .05 untuk kedua penelitian) dan tidak adanya gejala migrain yang paling mengganggu ( P = .05 untuk kedua penelitian). 17 , 18 Secara signifikan lebih banyak pasien yang menerima ubrogepant 50 dan 100 mg melaporkan tidak ada kecacatan ( P < .05), tingkat fungsi normal lebih besar ( P < .05), dan tingkat kemampuan fungsional normal yang dilaporkan peserta lebih tinggi ( P < .0001) dibandingkan dengan plasebo dari 2 hingga 8 jam setelah pemberian dosis. 19 Selain itu, 2 jam setelah pemberian dosis, lebih banyak pasien melaporkan perubahan keseluruhan dalam migrain mereka “jauh lebih baik” atau “secara signifikan lebih baik” dengan ubrogepant dibandingkan dengan plasebo ( P < .05). 19 Data gabungan dari kedua uji coba penting menunjukkan pemisahan ubrogepant 50 mg dan plasebo terjadi 1 jam setelah pemberian dosis untuk nyeri dan 1,5 jam untuk tidak adanya gejala yang paling mengganggu. 20 Setelah pemisahan tercapai, pemisahan dipertahankan selama 48 jam. 20 Dalam studi keamanan dan tolerabilitas dosis ganda yang mengevaluasi dosis supraterapeutik 150 mg sekali sehari selama 28 hari, berdasarkan pengukuran sistematis alanine aminotransferase serum, tidak ada tanda-tanda cedera hati akibat obat yang diamati. 13 Kejadian buruk klinis (AE) yang paling sering dilaporkan dalam studi Fase 1 meliputi nasofaringitis, sakit kepala, dan mual. 13 – 16 AE umumnya sementara dan intensitasnya ringan hingga sedang. 13 – 15 AE yang paling umum dalam studi Fase 3 adalah mual. 17 , 18Selain itu, tidak ada perbedaan yang relevan secara klinis dalam paparan sistemik dan tidak ada perbedaan dalam keamanan ketika ubrogepant diberikan kepada peserta Jepang yang sehat dibandingkan dengan peserta kulit putih. 21 Farmakokinetik proporsional dosis (PK) dicatat dalam rentang dosis 1-400 mg, dengan konsentrasi obat plasma maksimum (Cmax ) dicapai dalam 0,5-1,5 jam, 22 waktu paruh eliminasi terminal pendek (t 1/2 ; 5-7 jam), 13 , 16 dan tidak ada akumulasi setelah dosis berulang setiap hari. 13 Metabolisme ubrogepant terutama dimediasi melalui sitokrom P 450 (CYP) 3A4 dan eliminasi dimediasi melalui rute bilier dengan eliminasi ginjal minor. 12 , 23 Karena jalur eliminasi ini, penggunaan bersamaan inhibitor CYP3A4 yang kuat dikontraindikasikan, penggunaan penginduksi CYP3A4 yang kuat harus dihindari, dan inhibitor CYP3A4 sedang atau lemah mungkin memerlukan pengurangan dosis ubrogepant. 12 , 23 Namun, tidak ada perubahan signifikan dalam PK atau keamanan ketika ubrogepant diberikan bersamaan dengan antibodi monoklonal yang ditargetkan CGRP (erenumab dan galcanezumab) 24 atau atogepant, 25 yang digunakan untuk pengobatan pencegahan migrain pada orang dewasa. Ubrogepant adalah substrat protein resistensi kanker payudara dan transporter P-glikoprotein secara in vitro; oleh karena itu, penggunaan inhibitor protein resistensi kanker payudara dan/atau P-glikoprotein dapat meningkatkan paparan ubrogepant. Direkomendasikan agar pasien dengan gangguan hati atau ginjal berat menggunakan dosis awal 50 mg; ubrogepant tidak direkomendasikan untuk pasien dengan penyakit ginjal stadium akhir. 12 Ubrogepant merupakan substrat lemah dari transporter anion organik (OAT) P1B1, OATP1B3, dan OAT1. 23 Ubrogepant umumnya aman dan ditoleransi dengan baik dalam semua studi klinis.
Studi Tahap 1 ini dirancang untuk menetapkan bioekivalensi (BE) dari formulasi tablet 50 mg dan 100 mg yang akan dipasarkan (TBM) dengan formulasi tablet uji klinis (CT) yang diuji dalam uji coba Tahap 3 dan mengevaluasi efek makanan berlemak tinggi pada bioavailabilitas (BA) dari formulasi tablet TBM 100 mg.
Metode
Desain dan Penilaian Studi
Studi Fase 1 acak, dosis tunggal, label terbuka, terpusat tunggal ini dilakukan dalam 2 bagian. Bagian A adalah studi crossover 3 arah acak untuk menentukan BE dari 2 formulasi tablet TBM 50 mg dan 1 formulasi tablet CT 50 mg pada partisipan dewasa yang sehat. Perawatan diberikan dalam 1 dari 6 urutan pada Periode 1, 2, dan 3 dalam kondisi puasa dengan periode washout ≥7 hari di antara perawatan (Tabel 1 ). Bagian B adalah studi crossover 2 arah acak pada partisipan dewasa yang sehat untuk mengevaluasi efek makanan berlemak tinggi pada BA oral dari formulasi tablet TBM ubrogepant 100 mg. Perawatan diberikan dalam 1 dari 2 urutan pada Periode 1 dan 2 dengan periode washout ≥7 hari di antara perawatan (Tabel 1 ). Dosis 100 mg dipilih karena merupakan dosis klinis ubrogepant tertinggi yang dievaluasi dalam studi Fase 3.
Tabel 1. Perlakuan Studi

CT, uji klinis; TBM, akan dipasarkan.
Penelitian ini dilakukan sesuai dengan pedoman International Council for Harmonization E6 untuk Praktik Klinis yang Baik dan prinsip-prinsip Deklarasi Helsinki di Spaulding Clinical Research LLC (West Bend, WI). Semua partisipan memberikan persetujuan tertulis, dan peneliti memperoleh persetujuan protokol penelitian dari dewan peninjau institusional (Chesapeake Institutional Review Board, Columbia, MD) sebelum dimulainya penelitian.
Studi ubrogepant sebelumnya menemukan koefisien variasi dalam partisipan untuk C max dan area di bawah kurva konsentrasi plasma–waktu (AUC) sebesar 8%-16% dalam evaluasi dampak verapamil pada PK ubrogepant. 12 Dengan asumsi variabilitas intrasubjek sebesar 25%, ukuran sampel sekitar 42 partisipan dipilih untuk Bagian A untuk memastikan daya ≥90% bahwa interval kepercayaan 90% (CI) untuk rasio rata-rata geometrik uji/referensi (GMR) untuk parameter C max dan AUC berada dalam batas 80%-125%. Ukuran sampel 18 partisipan dipilih untuk Bagian B studi untuk memastikan penentuan yang andal tentang efek makanan pada BA oral ubrogepant dari formulasi tablet TBM.
Tujuan utama Bagian A adalah membandingkan paparan sistemik ubrogepant setelah pemberian dosis tunggal TBM dan formulasi tablet CT ubrogepant pada peserta dewasa yang sehat, dengan formulasi TBM sebagai variabel uji dan formulasi CT sebagai referensi (kontrol). Tujuan utama Bagian B adalah mengukur efek makanan berlemak tinggi pada paparan sistemik ubrogepant setelah pemberian dosis tunggal TBM formulasi tablet ubrogepant pada peserta dewasa yang sehat, dengan kondisi makan sebagai variabel uji dan kondisi berpuasa sebagai referensi (kontrol). Tujuan sekunder mencakup penilaian keamanan dan tolerabilitas ubrogepant yang diberikan sebagai formulasi tablet TBM atau CT.
Populasi Studi
Peserta di kedua bagian uji coba adalah pria dan wanita sehat berusia 18-45 tahun (inklusif), memiliki denyut nadi duduk ≥50 dan ≤100 denyut/menit, bukan perokok dan bukan pengguna nikotin, memiliki indeks massa tubuh ≥18 dan ≤30 kg/m 2 , tidak hamil, dan setuju untuk menggunakan metode kontrasepsi yang efektif.
Kriteria eksklusi meliputi penyakit apa pun yang signifikan secara klinis; kondisi klinis atau pembedahan apa pun yang akan membatasi penyerapan, distribusi, biotransformasi, atau ekskresi ubrogepant; riwayat penyalahgunaan alkohol atau zat lain dalam 5 tahun terakhir; hipersensitivitas terhadap antagonis reseptor CGRP, pengobatan sebelumnya atau bersamaan atau penggunaan obat bebas; hasil positif untuk obat-obatan terlarang; dan hasil elektrokardiogram abnormal atau hasil lab abnormal sebagaimana ditentukan oleh peneliti.
Analisis Farmakokinetik
Populasi PK terdiri dari semua peserta yang memiliki parameter PK yang dapat dievaluasi untuk ≥ 1 uji dan pengobatan referensi. Jika seorang peserta memiliki konsentrasi plasma ubrogepant sebelum dosis > 5% dari nilai C max dalam periode apa pun, data mereka dikecualikan untuk periode tersebut. Selain itu, jika ada peserta yang mengalami muntah pada atau sebelum 2 kali waktu konsentrasi obat plasma maksimum (t max ) untuk pengobatan saat kejadian tersebut terjadi, data periode untuk peserta dikecualikan.
Parameter PK untuk ubrogepant termasuk AUC dari waktu nol hingga waktu t (AUC 0–t ) dan dari waktu nol hingga tak terbatas (AUC 0–∞ ), C max , t max , konstanta laju eliminasi terminal, dan t 1/2 .
Sampel darah PK dikumpulkan untuk analisis ubrogepant plasma pada Hari ke-1, ke-8, dan ke-15 untuk Bagian A dan Hari ke-1 dan ke-8 untuk Bagian B pada 0 jam (sebelum pemberian dosis) dan 0,5, 1, 1,5, 2, 3, 4, 5, 6, 8, 12, 14, dan 24 jam setelah pemberian dosis pada setiap periode perawatan. Plasma diambil dari darah dalam waktu 30 menit setelah pengambilan darah, disentrifugasi pada > 2500 g selama 10 menit pada suhu sekitar 4°C, dipindahkan ke tabung polipropilena yang bersih, dibekukan dengan cepat, dan disimpan pada suhu -70°C hingga dianalisis. Konsentrasi ubrogepant dalam plasma ditentukan oleh Algorithme Pharma menggunakan metode kromatografi cair-spektrometri massa tandem yang tervalidasi dengan batas kuantisasi terendah 1,00 ng/mL dan rentang kalibrasi linier dari 1,00 hingga 1000 ng/mL. 14
Keamanan
Populasi keamanan mencakup semua peserta yang menerima ≥1 dosis pengobatan studi. Keamanan dinilai dengan memantau AE, uji laboratorium klinis, tanda-tanda vital, parameter elektrokardiogram, dan pemeriksaan fisik.
Analisis Farmakokinetik dan Statistik
Parameter PK nonkompartemen dihitung menggunakan Phoenix WinNonlin Versi 8.0 (Certara). Konsentrasi di bawah batas kuantifikasi ditetapkan ke nol. Data yang hilang tidak diperhitungkan. Semua analisis statistik dilakukan menggunakan SAS Versi 9.4 (SAS Institute). Statistik deskriptif parameter PK untuk ubrogepant disediakan untuk populasi PK berdasarkan perlakuan untuk setiap bagian penelitian. Parameter PK (Cmax , AUC0 –t , dan AUC0 –∞ ) dibandingkan menggunakan model efek campuran linear dengan urutan, perlakuan, dan periode sebagai efek tetap dan partisipan dalam urutan sebagai efek acak di setiap bagian penelitian. Untuk Bagian A dan B, rata-rata geometrik kuadrat terkecil dari setiap perlakuan ditentukan, dan 90% CI 2 sisi untuk rasio rata-rata geometrik kuadrat terkecil Cmax dan AUC antara perlakuan uji dan referensi dibuat. BE antara formulasi tablet TBM dan formulasi tablet CT dan tidak ada efek makanan pada PK disimpulkan jika GMR dan 90% CI yang sesuai berada dalam 80%-125%.
Hasil
Pada Bagian A, 47 peserta sehat didaftarkan, dengan usia rata-rata 32,3 tahun. Pada Bagian B, 18 peserta sehat didaftarkan, dengan usia rata-rata 28,4 tahun. Lihat Tabel S1 untuk informasi demografi terperinci. Konsentrasi pradosis ubrogepant berada di bawah batas kuantisasi pada setiap periode studi untuk semua peserta, yang menunjukkan pencucian yang cukup di antara perawatan.
Analisis Farmakokinetik
Bagian A
Paparan sistemik ubrogepant, berdasarkan Cmax , AUC 0–t , dan AUC 0–∞ , serupa setelah pemberian 1 × 100 mg dan 2 × 50 mg TBM (Perawatan A dan Perawatan B, berturut-turut) dan 2 × 50 mg CT (Perawatan C) di Bagian A (Tabel 2 dan Gambar 1A ). Selain itu, nilai t 1/2 rata-rata (sekitar 5-6 jam) serupa di seluruh perawatan, dan median tmax adalah 1 jam untuk setiap perawatan di Bagian A.
Tabel 2. Rata-rata (SD) Parameter PK Plasma Ubrogepant (Populasi PK)
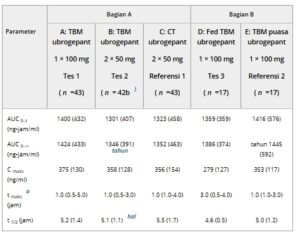
AUC 0–∞ , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga tak terhingga; AUC 0–t , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga waktu t; C max , konsentrasi obat plasma maksimum; CT, uji klinis; PK, farmakokinetik; SD, deviasi standar; t 1/2 , waktu paruh eliminasi terminal; t max , waktu konsentrasi obat plasma maksimum; TBM, akan dipasarkan.
Median (minimum–maksimum) dilaporkan untuk t maks .
b Kecuali dinyatakan sebaliknya.
c Meliputi data dari 41 peserta. AUC 0–∞ dikecualikan jika AUC yang diekstrapolasi >20%. Estimasi t 1/2 dihitung berdasarkan konstanta laju eliminasi terminal (λ z ). λ z ditentukan dengan melakukan analisis regresi pada fase linier terminal dari plot semilogaritmik data konsentrasi-waktu ubrogepant individual menggunakan minimal 3 titik konsentrasi-waktu dalam fase eliminasi, tidak termasuk C max . λ z dianggap valid jika r 2 > 0,8.
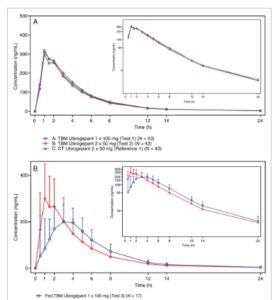
Gambar 1
Buka di penampil gambar
Kekuatan Gambar
Profil konsentrasi plasma rata-rata Ubrogepant–waktu untuk A) Bagian A, dan B) Bagian B (Populasi PK). CT, uji klinis (diproduksi di Merck, PA); TBM, akan dipasarkan (diproduksi di Allergan, Clonshaugh).
90% CI untuk GMR ubrogepant Cmax , AUC0 –t , dan AUC0 –∞ setelah pemberian Perawatan A dan Perawatan B versus Perawatan C di Bagian A berada dalam interval referensi 80%-125%; oleh karena itu, Perawatan A dan Perawatan B terbukti bioekuivalen dengan Perawatan C (Tabel 3 ). Selain itu, perbedaan median (uji-referensi) dalam waktu paparan puncak (tmax ) antara masing-masing perawatan TBM (Perawatan A dan B) dan Perawatan C adalah 0 jam, yang menunjukkan tmax sama untuk formulasi TBM dan CT.
Tabel 3. Ringkasan Perbandingan
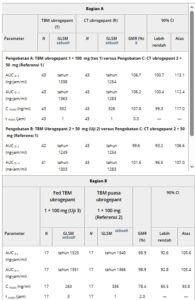
AUC 0–∞ , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga tak terhingga; AUC 0–t , area di bawah kurva konsentrasi plasma–waktu dari waktu nol hingga waktu t; C max , konsentrasi obat plasma maksimum; CT, uji klinis; GLSM, rata-rata kuadrat terkecil geometris; GMR, rasio rata-rata geometris; R, referensi; T, uji; t max , waktu konsentrasi obat plasma maksimum; TBM, yang akan dipasarkan.
Median dilaporkan untuk t maks .
b Perbedaan median (uji–referensi) dilaporkan untuk t maks .
c Perbedaan median mencakup data dari 42 peserta.
Kotak diagram parameter plasma ubrogepant PK untuk Bagian A menunjukkan nilai rata-rata dan median yang sebanding serta tumpang tindih yang cukup besar dalam rentang interkuartil di seluruh perlakuan untuk nilai C max , AUC 0–t , dan AUC 0–∞ untuk Perlakuan A dan B relatif terhadap Perlakuan C (Gambar 2A, B ). Pengamatan ini konsisten dengan perbandingan statistik parameter ubrogepant PK untuk Bagian A (Tabel 3 ).

Gambar 2
Buka di penampil gambar
Kekuatan Gambar
Kotak diagram parameter PK plasma ubrogepant untuk populasi PK untuk Bagian A (AB) dan Bagian B (CD). Berlian menunjukkan rata-rata, sedangkan tepi bawah dan atas kotak masing-masing mewakili kuartil pertama dan ketiga. Median diwakili oleh garis di dalam kotak. Kumis (garis yang memanjang dari kedua ujung kotak) mencapai hingga 1,5 kali rentang interkuartil. Titik data yang berada di luar rentang ini ditampilkan secara individual dan dianggap sebagai outlier potensial. CT, uji klinis (diproduksi di Merck, PA); TBM, akan dipasarkan (diproduksi di Allergan, Clonshaugh)
Bagian B
Profil konsentrasi-waktu rata-rata untuk ubrogepant plasma setelah pemberian dosis tunggal dari formulasi tablet TBM 100 mg dalam kondisi makan (Perlakuan D) dan puasa (Perlakuan E) ditunjukkan pada Gambar 1B .
Paparan sistemik terhadap ubrogepant (berdasarkan AUC 0–t dan AUC 0–∞ ) serupa setelah pemberian formulasi TBM dalam kondisi kenyang dan puasa; namun, konsentrasi plasma puncak rata-rata (Cmax ) lebih rendah, dan median tmax tertunda (dari 1 hingga 3 jam) dalam kondisi kenyang dibandingkan dengan kondisi puasa (Tabel 2 ). Konsisten dengan Bagian A, t 1/2 rata-rata adalah sekitar 5 jam untuk kedua perawatan di Bagian B.
Perbandingan statistik parameter PK ubrogepant setelah pemberian formulasi tablet TBM 100 mg dalam kondisi diberi makan dibandingkan dengan kondisi berpuasa (Perlakuan D vs Perlakuan E) di Bagian B disajikan dalam Tabel 3. 90% CI untuk GMR AUC 0-t ubrogepant dan AUC 0-∞ untuk perlakuan diberi makan versus berpuasa berada dalam interval referensi 80%-125%; oleh karena itu, makanan tidak memengaruhi tingkat paparan ubrogepant untuk formulasi tablet TBM 100 mg. Namun, 90% CI untuk GMR C max berada di luar interval referensi 80%-125%. Berdasarkan GMR, makanan menurunkan paparan puncak ubrogepant sekitar 22% untuk formulasi tablet TBM 100 mg. Selain itu, perbedaan (makan-puasa) dalam median t max antara perlakuan adalah 2 jam, yang menunjukkan bahwa makanan berlemak tinggi menunda t max setelah pemberian formulasi tablet TBM 100 mg. Boxplot parameter AUC ubrogepant plasma untuk Bagian B menunjukkan nilai rata-rata dan median yang sebanding dan tumpang tindih yang cukup besar dalam rentang interkuartil antara perlakuan untuk formulasi tablet TBM 1 × 100 mg yang diberikan dalam kondisi diberi makan dibandingkan dengan kondisi puasa (Gambar 2C, D ). Namun, untuk C max , tidak banyak tumpang tindih dalam rentang interkuartil antara 2 perlakuan, dan nilai rata-rata dan median lebih rendah untuk formulasi tablet TBM 1 × 100 mg yang diberikan dalam kondisi diberi makan dibandingkan dengan kondisi puasa. Pengamatan ini konsisten dengan perbandingan statistik parameter PK ubrogepant untuk Bagian B (Tabel 3 ).
Keamanan
AE yang muncul akibat pengobatan (TEAE) dirangkum dalam Tabel S2 . Persentase keseluruhan peserta yang mengalami TEAE dan TEAE terkait pengobatan rendah dan sebanding di antara kelompok pengobatan. Tidak ada kematian atau TEAE parah atau serius yang dilaporkan baik di Bagian A maupun Bagian B dari penelitian ini.
Perubahan rata-rata nilai laboratorium klinis dari awal berada dalam variasi normal dan tidak dianggap bermakna secara klinis. Demikian pula, tidak ada tren bermakna secara klinis yang diamati dalam rata-rata atau perubahan dari awal dalam pengukuran tanda vital atau hasil elektrokardiogram.
Diskusi
Studi Fase 1 crossover, berlabel terbuka, 2 bagian ini memungkinkan setiap peserta untuk bertindak sebagai kontrol mereka sendiri, meningkatkan ketepatan estimasi perbedaan pengobatan dan mengurangi jumlah keseluruhan peserta yang diperlukan untuk penelitian ini. Bagian A dari penelitian menemukan bahwa dosis oral tunggal dari 2 kekuatan yang diuji (1×100 mg atau 2×50 mg) dari formulasi tablet TBM ubrogepant adalah bioekuivalen dengan formulasi tablet CT (2×50 mg) ketika diberikan dalam kondisi puasa pada orang dewasa yang sehat. Selain itu, waktu rata-rata untuk paparan maksimum (1 jam) dan rata-rata t 1/2 sebanding di seluruh perawatan di Bagian A. Bagian B menemukan bahwa makanan berlemak tinggi tidak memengaruhi tingkat paparan sistemik ubrogepant (berdasarkan AUC 0–t dan AUC 0–∞ ) setelah pemberian dosis tunggal tablet TBM 100 mg; namun, C max sekitar 22% lebih rendah dalam kondisi makan dibandingkan dengan kondisi puasa. T maks setelah pemberian tablet TBM 100 mg tertunda dalam kondisi kenyang (dengan perbedaan median 2 jam), yang menunjukkan menurunnya laju penyerapan akibat makanan berlemak tinggi.
Idealnya, suatu obat akan memiliki profil PK yang sama dalam kondisi kenyang dan puasa. Namun, diketahui secara umum bahwa asupan makanan dapat memengaruhi profil PK suatu obat karena berbagai perubahan fisiologis pasca makan yang terjadi, termasuk perubahan volume cairan, pH, motilitas, waktu tinggal lambung, dan potensi penyerapan usus yang tertunda. 26 , 27 Lebih jauh lagi, kelarutan rendah dan permeabilitas rendah ubrogepant mempersulit dampak makanan pada pelarutan obat secara in vivo dan BA, yang mengarah pada hasil yang tidak dapat diprediksi tentang bagaimana makanan dapat memengaruhi penyerapan obat di saluran pencernaan. 28 , 29 C max Ubrogepant menurun, t max tertunda, dan penyerapan keseluruhan tidak terpengaruh setelah makan berlemak tinggi. Sementara t max yang cepat dan C max yang lebih tinggi diyakini berkontribusi pada onset aksi yang cepat dan penyembuhan migrain yang cepat, ubrogepant adalah antagonis reseptor CGRP yang poten (Ki = 0,07 nM) dengan konsentrasi yang menghasilkan respons maksimal 90% (13 ng/mL) konsentrasi plasma berdasarkan model vasodilatasi dermal yang diinduksi capsaicin manusia yang dicapai dalam waktu 11 menit dalam studi pivotal di mana obat diberikan tanpa batasan makanan apa pun. 20 Dalam salah satu studi pivotal, dosis ubrogepant 25 mg yang rendah (tidak disetujui) juga menunjukkan kebebasan nyeri yang signifikan pada 2 jam dibandingkan dengan plasebo. 18 Selain itu, untuk pengobatan akut serangan migrain, pasien tidak dapat merencanakan kapan harus minum obat; sebaliknya, mereka harus minum obat saat dibutuhkan, terlepas dari status makanan mereka. Dengan demikian, efek makanan yang diamati tidak dianggap relevan secara klinis, dan obat tersebut direkomendasikan untuk diminum tanpa memperhatikan makanan.
Tidak ada perbedaan yang relevan secara klinis dalam keamanan atau tolerabilitas formulasi tablet TBM ubrogepant (kekuatan 50 dan 100 mg) dan formulasi tablet CT (kekuatan 50 mg) yang diamati dalam kondisi puasa (Bagian A) atau setelah pemberian tablet TBM 100 mg dalam kondisi makan (Bagian B). Insiden TEAE secara keseluruhan rendah selama kedua bagian penelitian (masing-masing 19,1% dan 11,1% untuk Bagian A dan Bagian B), dan tidak ada TEAE parah atau serius yang dilaporkan. Tidak ada perubahan yang relevan secara klinis dalam nilai laboratorium klinis, tanda-tanda vital, atau parameter elektrokardiogram.
Kesimpulan
Dosis oral tunggal tablet TBM ubrogepant 100 mg (1×100 mg atau 2×50 mg) terbukti bioekuivalen dengan tablet CT saat diberikan dalam kondisi puasa. Tidak ada efek klinis yang relevan dari makanan berlemak tinggi pada PK tablet TBM ubrogepant. Baik tablet TBM maupun CT yang diuji aman dan dapat ditoleransi dengan baik saat diberikan dalam kondisi puasa dan/atau makan kepada peserta dewasa yang sehat.

